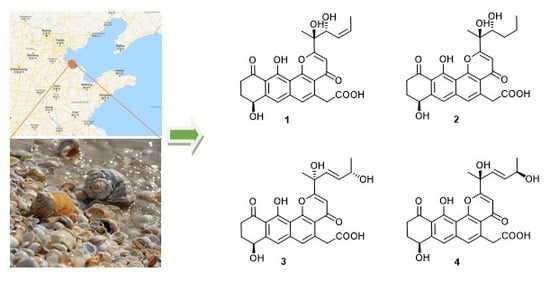
Compound
1 was isolated as a yellow oil, and a yellow crystal was obtained by recrystallization with methanol/acetone (5:1) mixed solvent. The high resolution ESI-MS gave a quasi-molecular ion at
m/
z 469.1493 [M + H]
+ (
Figure S1), which is consistent with the molecular formula C
25H
24O
9 (calcd. for C
25H
25O
9+, 469.1493). The NMR spectra of
1 (
Figures S2–S7 and
Table 1) displayed 12 signals, including two methyls, three methylenes, three olefinic protons, and two aromatic protons. The
13C-NMR spectroscopic data of
1 showed three carbonyl signals at δ
C 175.7, 182.1, and 185.4, and eight quaternary carbon signals (δ
C 165.8, 159.1, 147.8, 142.6, 138.0, 120.6, 114.7, and 113.3), which are characteristics for the existence of the ring-fused aromatic system. The presence of
1H-
1H spin–spin coupling system in the COSY experiment of
1 from H-8 to H-9 and H-9 to H-10 combined with the HMBC correlations from H-10 to C-11 and C-11a, and another HMBC correlations from H-8 to C-7a and C-7 confirmed the structure of ring A. The chemical shift of H-8 was as high as 4.95 ppm indicated that it was connected with a hydroxyl group. Two aromatic proton signals suggested that this compound has a multi-substituted naphthalene structure. Additionally, the HMBC correlations from H-7 to C-8, C-6 and C-11a combined with the HMBC correlations from H-6 to C-7, C-13, C12a, C-4a revealed the substitution of naphthalene rings. More in-depth, the chemical shift of C-12 moved to the downfield at 165.8 ppm, suggesting that the C-12 was connected with an oxygen atom. Then, the structure of ring B, C, and D was confirmed. The distinct signals of H-13 (δ
H 4.16, 4.26) and the HMBC correlations from this nucleus to C-13-COOH, C-6, and C-4a gave the information that the carboxymethyl group was connected with C-5 on the naphthalene core. The γ-pyrone structure (ring A) formed by C-2, C-3, C-4, C-4a, and C-12b was also deduced by the HMBC correlations from H-3 to C-2, C-4, C-4a. According to the
1H-
1H COSY of compound
1, there was still a very significant spin-spin coupling system from H-19 to H-18 then to H17 and stopped at H-16. The chemical shift of H-17 (5.73 ppm) and H-18 (5.88 ppm) suggested an ethylenic bond between C-17 and C-18. It was clear to verify the geometry of the C-17/18 double bond is
Z according to the
cis-
1H–
1H coupling between H-17 and H-18 (
J = 11.7 Hz) and the single crystal X-ray diffraction. The side chain structure was elucidated by HMBC correlations from H-17 to C-16, H-16 to C-2, C-3, and C-15 combined with the HMBC correlations from H-15 to C-2, C-3, C-16, and from H-3 to C-14. Then the planar structure of compound
1 was illustrated. As shown in
Figure 2, the NOESY cross-peaks from H-3 to H-15, H-15 to H-16, and H-15 to H-17 (
Figure 2, mixing time 700 ms) were observed but that cannot give definite evidence to support the 14
S*, 16
R* relative configuration. Compound
1 can form fine crystals in methanol and acetone mixtures, so the single crystal X-ray diffraction was tested, and the absolute configuration of compound
1 was determined (
Figure 3, Crystal structure reports see
Supplementary Materials; CCDC deposition number: 1970563). Based on the above analyses, the gross structure of
1 was deduced as a new polyketide, for which we proposed the name shellmycin A.
Compound
2 was isolated as a yellow oil. The molecular formula of
2 was assigned as C
25H
26O
9 by interpretation of a protonated molecular ion peak at
m/
z 471.1648 [M + H]
+ (calcd. for C
25H
27O
9+, 471.1650) in the HR-MS spectrum (
Figure S8). Compared to
1, compound
2 has two more hydrogen atoms. The NMR data (
Figures S9–S14) demonstrated that compound 2 has a multi-substituted naphthalene structure (
Table 2), just like
1. The same noticeable
1H-
1H COSY cross-peaks from H-10 to H-9 and H-9 to H-8 additional with the HMBC correlation from H-10 to C-11a and H-8 to C-7 suggested the structure of the ring A is the same as compound
1. Similarly, the HMBC correlation from H-7 to C-8, C-6, and another HMBC correlation from H-6 to C-7, C-13, and C-4a as well as the HMBC correlation from H-13 to C-6 and C-4a suggested that the compound
2 has the same naphthalene core with
1 (
Figure 4). Similarly, the HMBC correlation from H-3 to C-4a, C-14, and C-2 could also ascertain the γ-pyrone structure, and its substitution type was the same as
1. The apparent difference of the
1H-NMR and
13C-NMR between
2 and
1 was that the characteristic peaks of the double bond disappeared. Distinct
1H-
1H COSY cross peaks from H-16 to H-17 then from H-17 to H-18 then to H-19, and their HMBC correlations gave the evidence, which confirmed the side chain structure of compound
2. Same as
1, the NOESY cross-peaks from H-3 to H-15, H-15 to H-16, and H-15 to H-17 cannot give definite evidence to support the 14
S*, 16
R* relative configuration. The 8S* relative configuration was confirmed from the single-crystal structure of 1 and proposed biosynthetic pathways. Based on comparison of the ECD spectra between
1 and
2, the absolute configuration of
2 was assigned (
Figure 5). Thus, the structure of shellmycin B (
2) was proposed.
The molecular formula of
3 and
4 were both assigned as C
25H
26O
9 by interpretation of a protonated molecular ion peak at
m/
z 469.1491 and 469.1494 (calcd for C
25H
25O
9+, 469.1493) in the HR-MS spectrum (
Figures S15 and S22). Interestingly, these two compounds had almost identical NMR spectra. These facts showed that compound
1 and
4 have the same planar structure. The comparison of its
1H and
13C NMR data (
Table 3 and
Table 4,
Figures S16–S21;
Figures S23–S28) with those of
1 indicated that both structures contain the same backbone fused with the γ-pyrone and naphthalene ring. The
1H-
1H COSY from H-19 to H-18 (δ
H 4.32) gave the evidence that C-18 is connected with a hydroxy-substituted carbon (C18, δ
C 69.4). Then the four-carbon chain from C-16 to C-19 was established according to
1H–
1H COSY correlations, along with the HMBC correlations from the protons of H-16, H-17, H-18, and H-19 to the corresponding carbons (
Figure 6). The geometry of the C-16/17 double bond was determined to be
E-form based on the
trans-
1H–
1H coupling constants between H-16 and H-17 (
J = 15.5 Hz). The 8
S* relative configuration was proposed based on analogy to the single-crystal structure of
1 and proposed biosynthetic pathways. Most impressive of all, the
3 and
4 have the same planar structure but difference retention time between
3 and
4 is likely due to the stereochemistry of C-14 and C-18. In compound
4, a distinct NOESY cross peak was found between H-15 and H-16, but there was not any signal between H-15 and H-17. A very different NOESY correlation from H-15 to both H-16 and H-17 suggested that there have some difference between
3 and
4. Based on the ECD spectra and calculated spectra of
3 and
4, the absolute configurations of
3 and
4 were assigned (
Figure 7) that the stereochemistry of C-14 should be
R* in
4 but
S* in
3. Thus,
3 and
4 are a pair of stereoisomers, called shellmycin C (
3) and shellmycin D (
4).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}