Heteroexpression of Aspergillus nidulans laeA in Marine-Derived Fungi Triggers Upregulation of Secondary Metabolite Biosynthetic Genes

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Bioinformatics Analysis of Az5LaeA and AnLaeA
2.2. Overexpression of laeA Genes
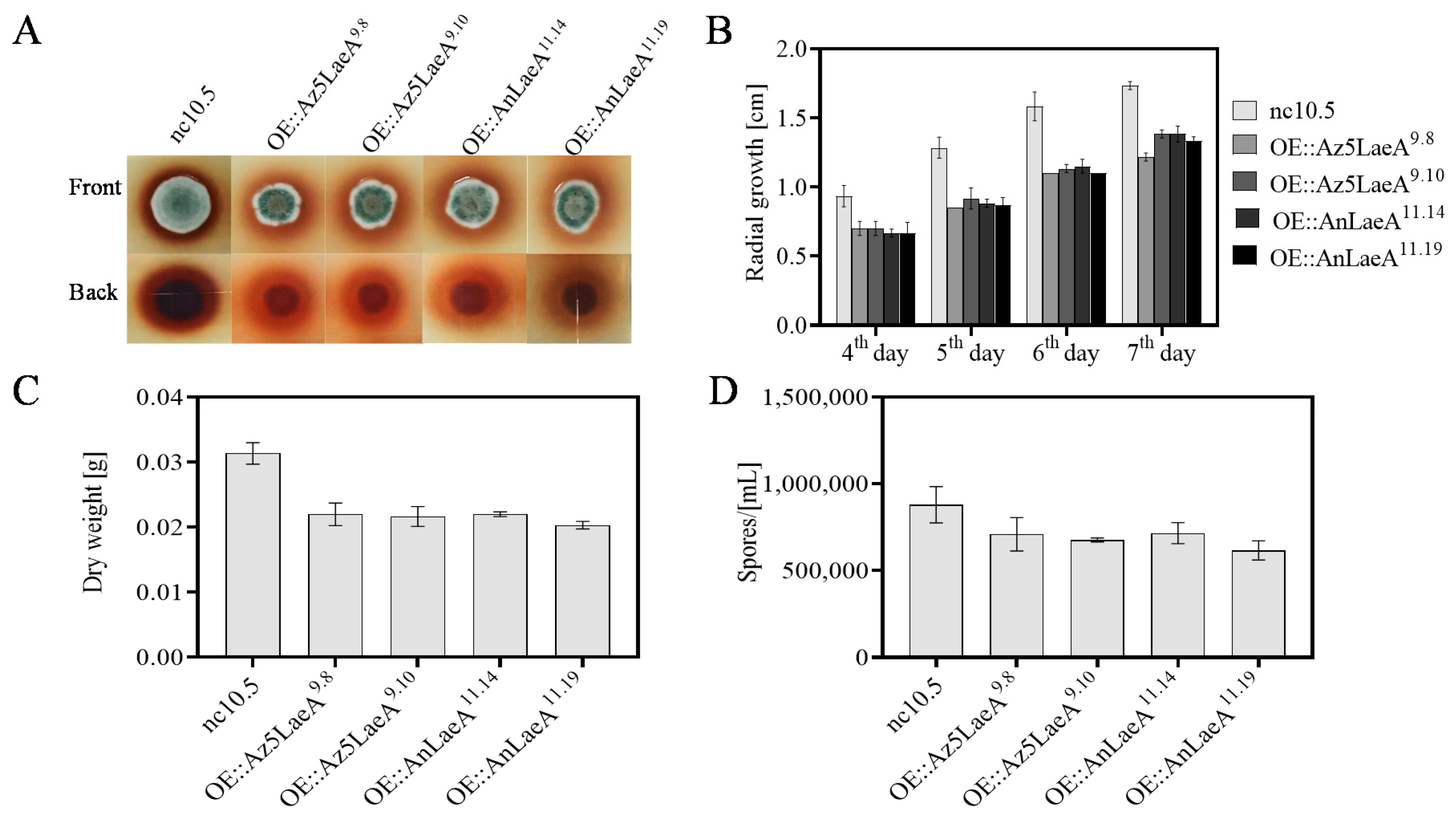
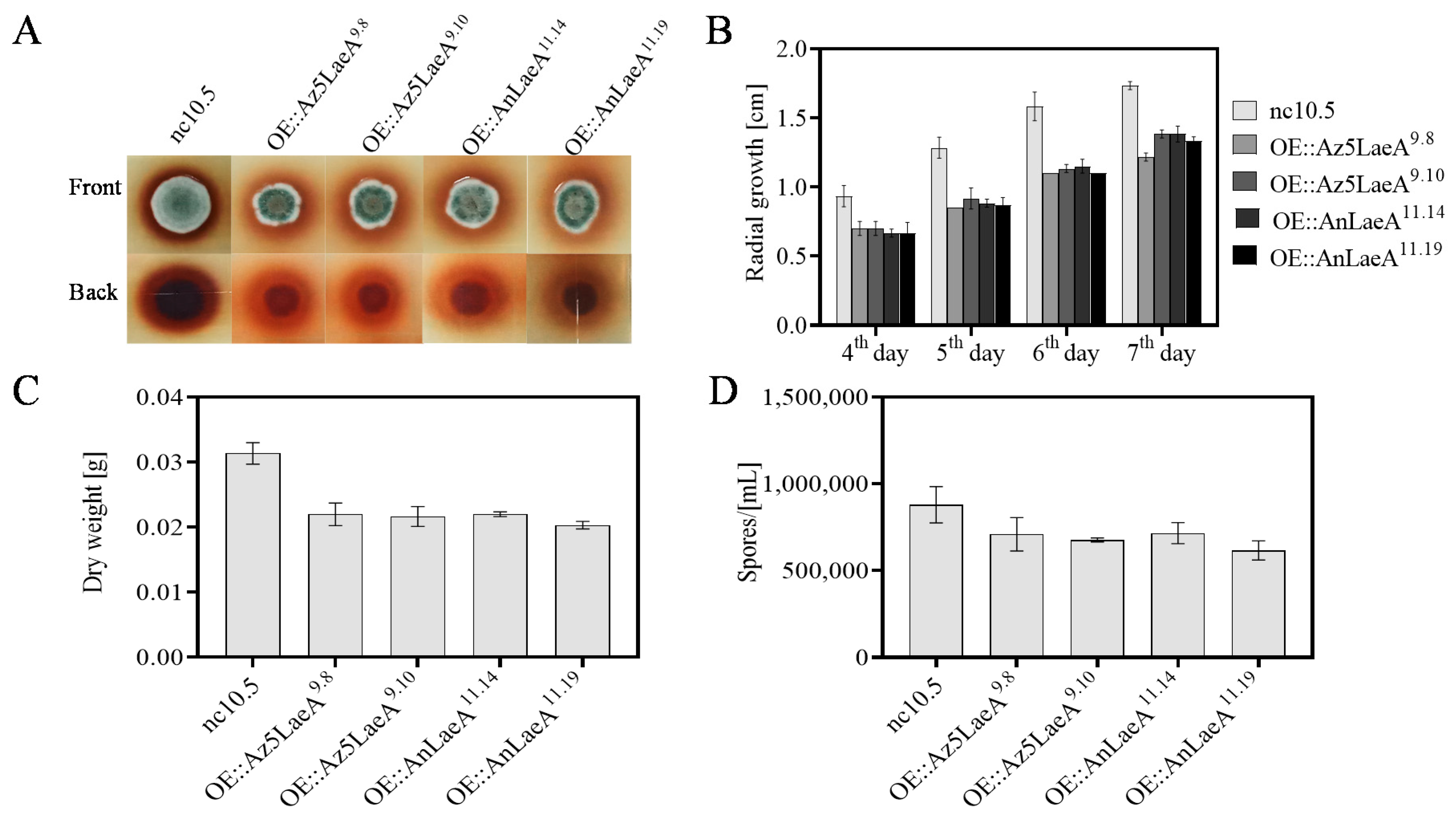
2.3. Phenotype and Scanning Electron Microscope Analysis
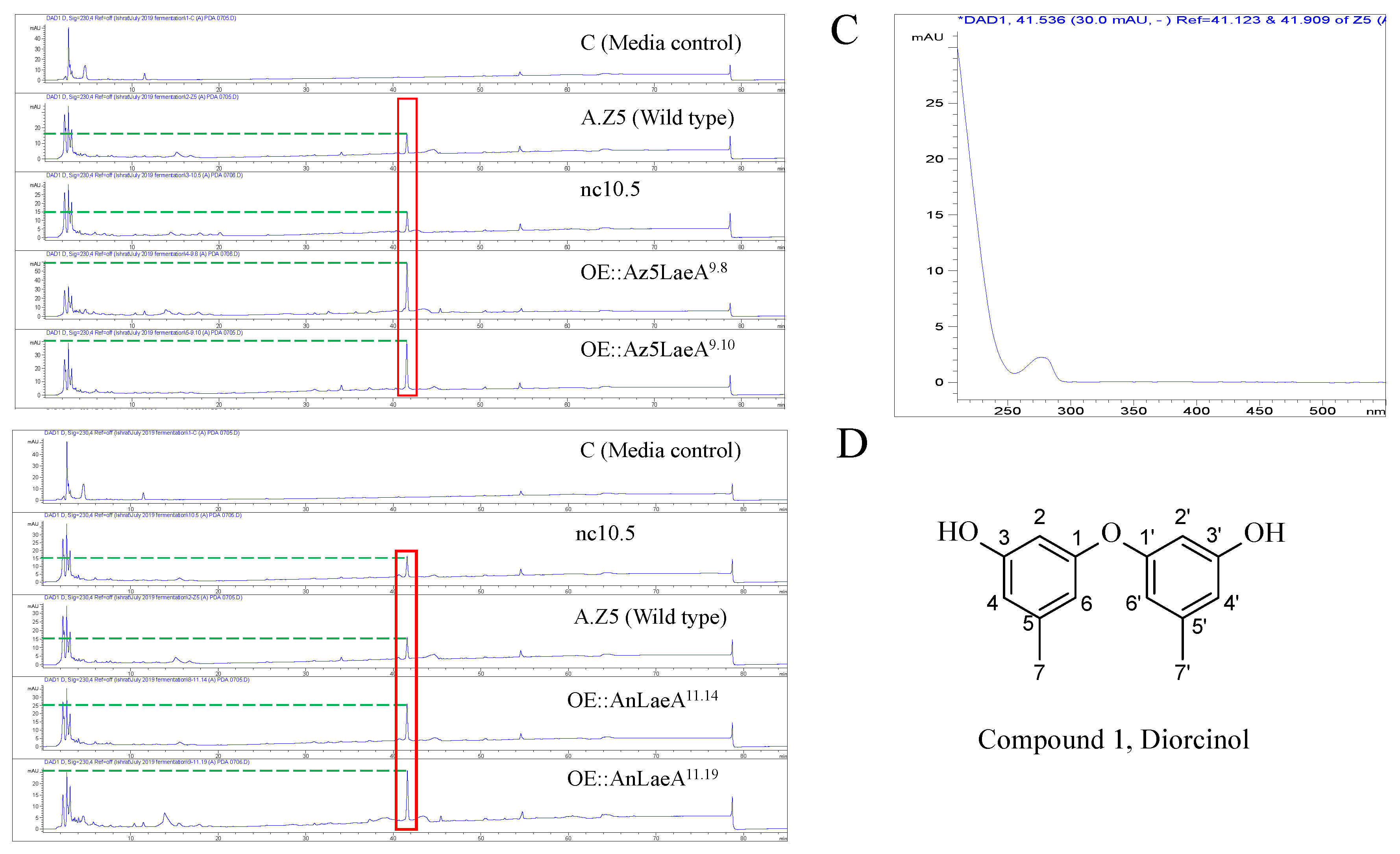
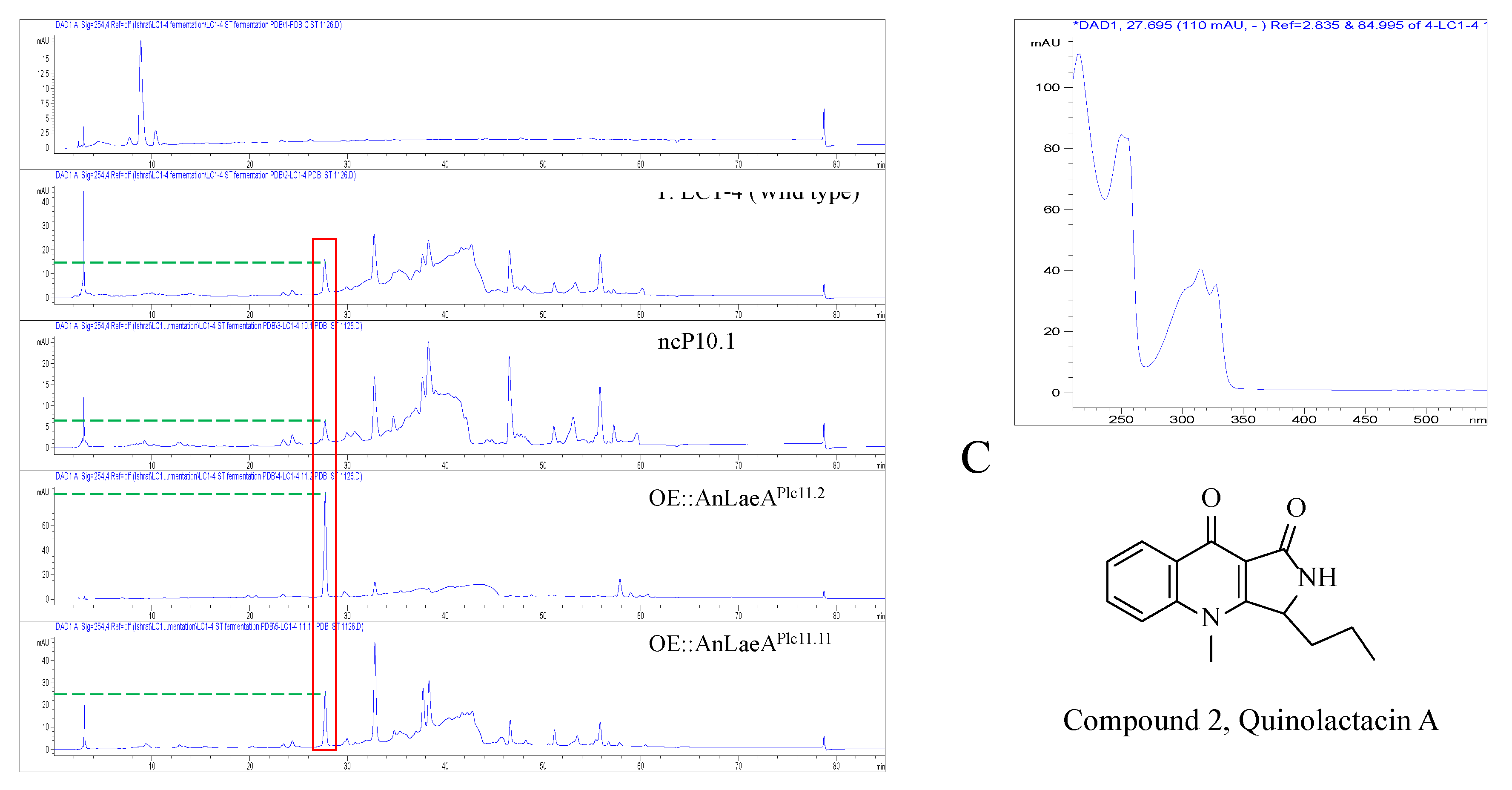
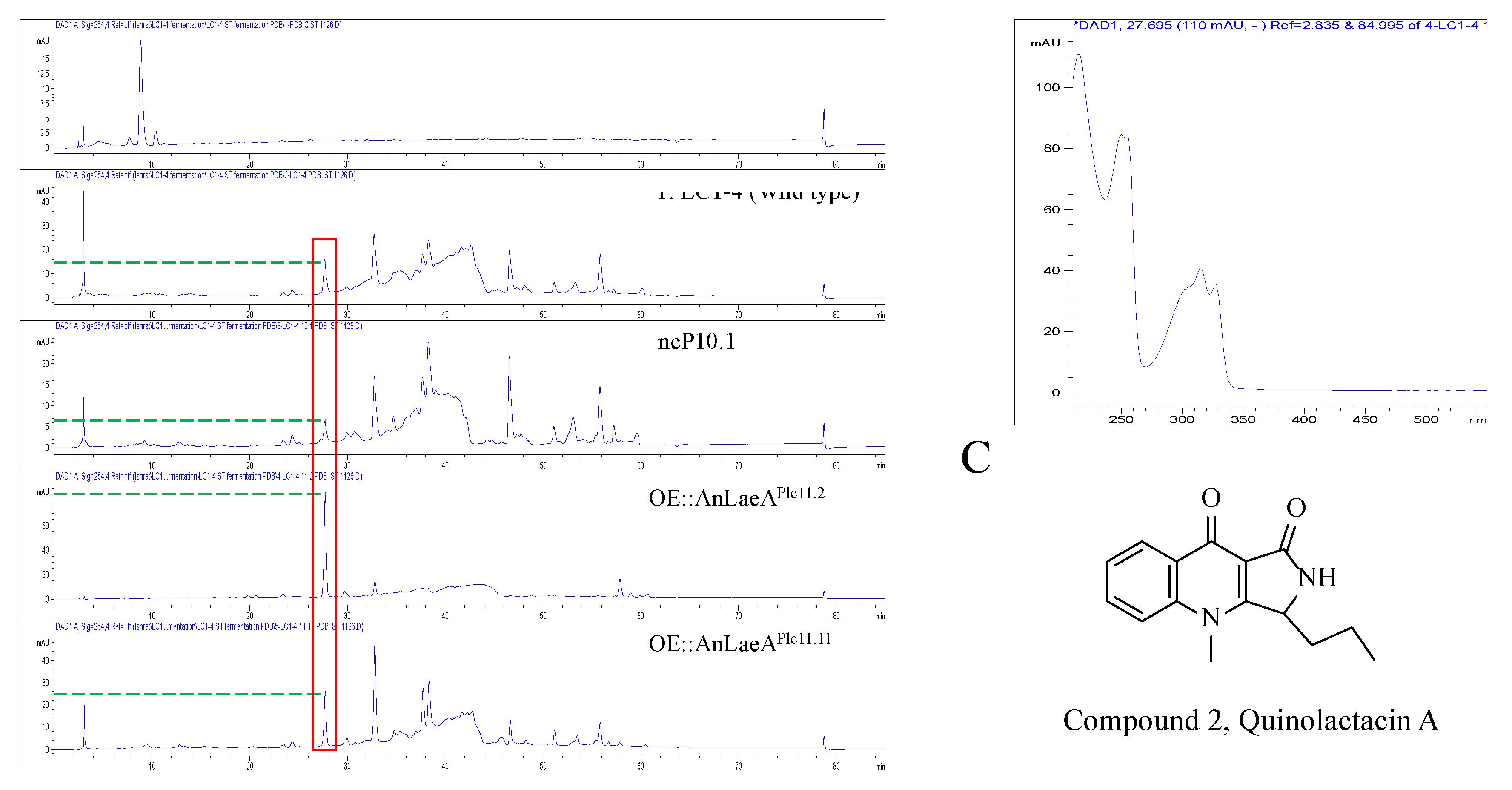
2.4. Secondary Metabolites Analysis
2.5. Transcriptome Analysis
2.6. Functional Annotation and Differential Gene Expression of Unigenes
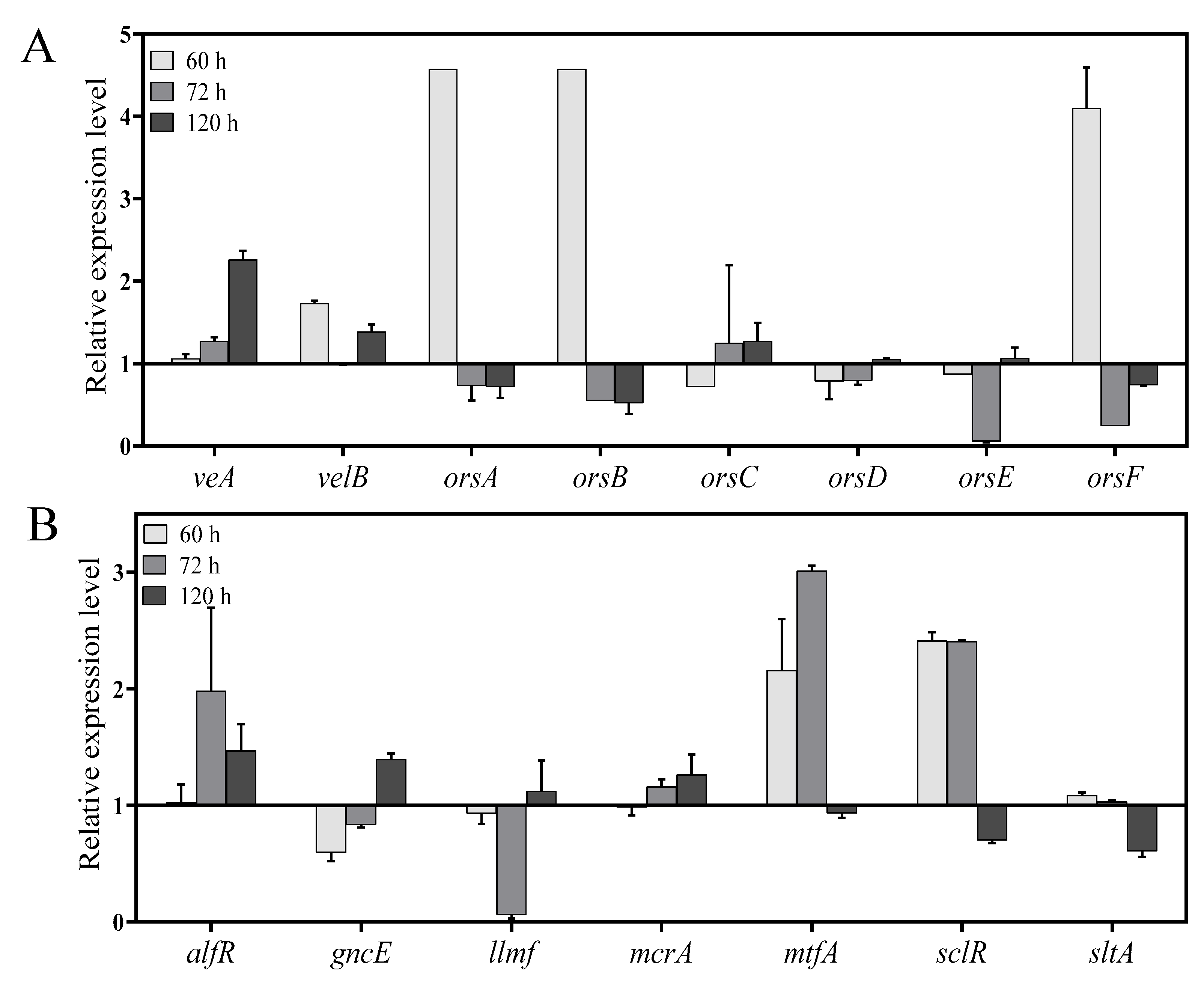
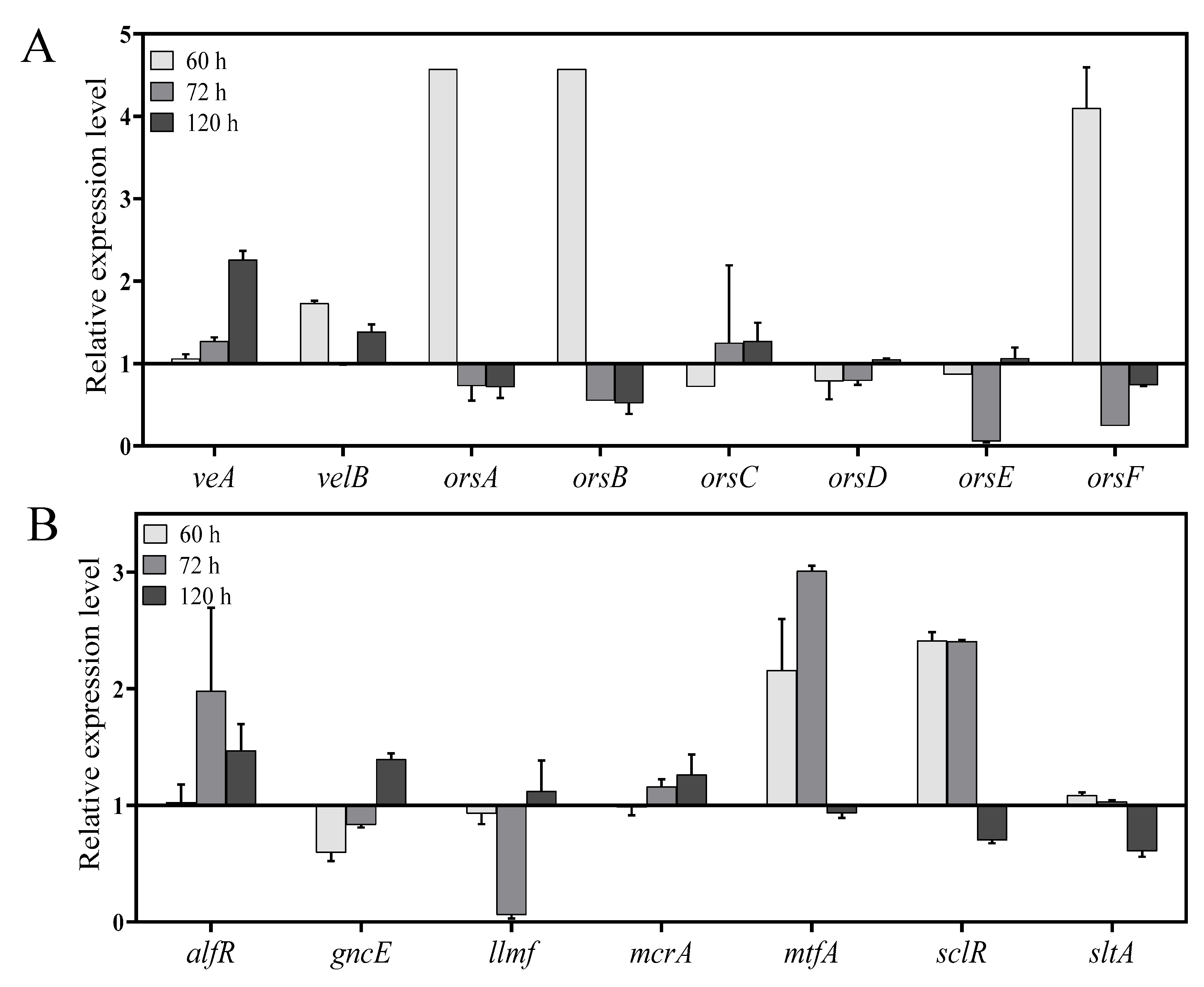
2.7. In Vitro Validation of Specific Genes by qPCR
3. Discussion
4. Materials and Methods
4.1. Strains, Media, and Culture Conditions
4.2. Gene Cloning and Bioinformatics Analysis
4.3. Molecular Genetic Manipulations
4.4. Fungal Transformation
4.5. Phenotype Analysis and Scanning Electron Microscopy
4.6. Secondary Metabolite Analysis, Purification, and Identification of Compound
4.7. Transcriptome Analysis
4.8. In Vitro Validation of Specific Genes by qPCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kong, F.; Wang, Y.; Liu, P.; Dong, T.; Zhu, W. Thiodiketopiperazines from the Marine-Derived Fungus Phoma sp. OUCMDZ-1847. J. Nat. Prod. 2014, 77, 132–137. [Google Scholar] [CrossRef] [PubMed]
- An, C.-Y.; Li, X.-M.; Luo, H.; Li, C.-S.; Wang, M.-H.; Xu, G.-M.; Wang, B.-G. 4-Phenyl-3,4-Dihydroquinolone Derivatives from Aspergillus Nidulans MA-143, an Endophytic Fungus Isolated from the Mangrove Plant Rhizophora Stylosa. J. Nat. Prod. 2013, 76, 1896–1901. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhang, X.; Du, L.; Wang, W.; Zhu, T.; Gu, Q.; Li, D. Sorbicatechols A and B, Antiviral Sorbicillinoids from the Marine-Derived Fungus Penicillium Chrysogenum PJX-17. J. Nat. Prod. 2014, 77, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [Green Version]
- Rateb, M.E.; Ebel, R. Secondary Metabolites of Fungi from Marine Habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Lyu, H.-N.; Liu, H.-W.; Keller, N.; Yin, W.-B. Harnessing Diverse Transcriptional Regulators for Natural Product Discovery in Fungi. Nat. Prod. Rep. 2019, 37. [Google Scholar] [CrossRef]
- Brakhage, A.A.; Schuemann, J.; Bergmann, S.; Scherlach, K.; Schroeckh, V.; Hertweck, C. Activation of Fungal Silent Gene Clusters: A New Avenue to Drug Discovery. Prog. Drug. Res. 2008, 66, 3–12. [Google Scholar] [CrossRef]
- Netzker, T.; Fischer, J.; Weber, J.; Mattern, D.J.; König, C.C.; Valiante, V.; Schroeckh, V.; Brakhage, A.A. Microbial Communication Leading to the Activation of Silent Fungal Secondary Metabolite Gene Clusters. Front. Microbiol. 2015, 6, 299. [Google Scholar] [CrossRef]
- Rutledge, P.J.; Challis, G.L. Discovery of Microbial Natural Products by Activation of Silent Biosynthetic Gene Clusters. Nat. Rev. Microbiol. 2015, 13, 509–523. [Google Scholar] [CrossRef]
- Bok, J.W.; Keller, N.P. LaeA, A Regulator of Secondary Metabolism in Aspergillus spp. Eukaryot. Cell 2004, 3, 527–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Lee, J.-B.; Lee, I. Strain Improvement by Overexpression of the laeA Gene in Monascus Pilosus for the Production of Monascus-Fermented Rice. J. Microbiol. Biotechnol. 2013, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Wang, M.; Li, L.; Si, J.; Song, B.; Zhou, C.; Yu, M.; Wang, X.; Zhang, Y.; Ding, G.; et al. Overexpression of the Global Regulator LaeA in Chaetomium Globosum Leads to the Biosynthesis of Chaetoglobosin Z. J. Nat. Prod. 2016, 79, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Chettri, P.; Bradshaw, R.E. LaeA Negatively Regulates Dothistromin Production in the Pine Needle Pathogen Dothistroma Septosporum. Fungal Genet. Biol. 2016, 97, 24–32. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, H.; Zhu, Q.; Hao, S.; Chai, S.; Li, Y.; Jiao, Z.; Shi, J.; Sun, B.; Wang, C. Overexpression of Global Regulator LaeA Increases Secondary Metabolite Production in Monascus Purpureus. Appl Microbiol Biotechnol. 2020, 104, 3049–3060. [Google Scholar] [CrossRef]
- Hong, E.J.; Kim, N.K.; Lee, D.; Kim, W.G.; Lee, I. Overexpression of the LaeA Gene Leads to Increased Production of Cyclopiazonic Acid in Aspergillus Fumisynnematus. Fungal Biol. 2015, 119, 973–983. [Google Scholar] [CrossRef]
- Yu, J.; Han, H.; Zhang, X.; Ma, C.; Sun, C.; Che, Q.; Gu, Q.; Zhu, T.; Zhang, G.; Li, D. Discovery of Two New Sorbicillinoids by Overexpression of the Global Regulator LaeA in a Marine-Derived Fungus Penicillium Dipodomyis YJ-11. Mar. Drugs 2019, 17, 446. [Google Scholar] [CrossRef] [Green Version]
- Bok, J.W.; Noordermeer, D.; Kale, S.P.; Keller, N.P. Secondary Metabolic Gene Cluster Silencing in Aspergillus Nidulans. Mol. Microbiol. 2006, 61, 1636–1645. [Google Scholar] [CrossRef]
- Bok, J.W.; Hoffmeister, D.; Maggio-Hall, L.A.; Murillo, R.; Glasner, J.D.; Keller, N.P. Genomic Mining for Aspergillus Natural Products. Chem. Biol. 2006, 13, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Bouhired, S.; Weber, M.; Kempf-Sontag, A.; Keller, N.P.; Hoffmeister, D. Accurate Prediction of the Aspergillus Nidulans Terrequinone Gene Cluster Boundaries Using the Transcriptional Regulator LaeA. Fungal. Genet. Biol. 2007, 44, 1134–1145. [Google Scholar] [CrossRef]
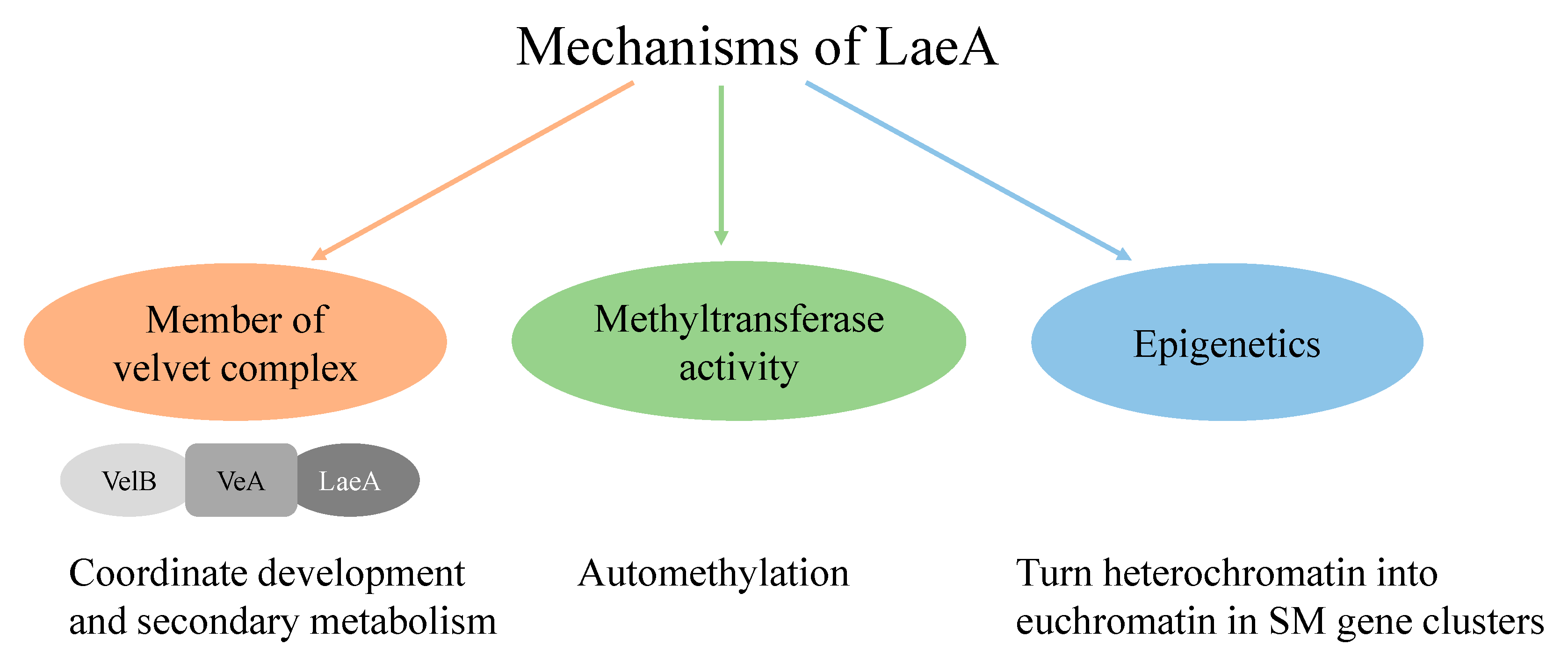
- Patananan, A.N.; Palmer, J.M.; Garvey, G.S.; Keller, N.P.; Clarke, S.G. A Novel Automethylation Reaction in the Aspergillus Nidulans LaeA Protein Generates S-Methylmethionine. J. Biol. Chem. 2013, 288, 14032–14045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Dominguez, Y.; Bok, J.W.; Berger, H.; Shwab, E.K.; Basheer, A.; Gallmetzer, A.; Scazzocchio, C.; Keller, N.; Strauss, J. Heterochromatic Marks Are Associated with the Repression of Secondary Metabolism Clusters in Aspergillus Nidulans. Mol. Microbiol. 2010, 76, 1376–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayram, O.; Krappmann, S.; Ni, M.; Bok, J.W.; Helmstaedt, K.; Valerius, O.; Braus-Stromeyer, S.; Kwon, N.J.; Keller, N.P.; Yu, J.H.; et al. VelB/VeA/LaeA Complex Coordinates Light Signal with Fungal Development and Secondary Metabolism. Science 2008, 320, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; Johnstone, I.L.; Clutterbuck, A.J. An Autonomously Replicating Plasmid Transforms Aspergillus Nidulans at High Frequency. Gene 1991, 98, 61–67. [Google Scholar] [CrossRef]
- Aleksenko, A.; Clutterbuck, A.J. The Plasmid Replicator AMA1 in Aspergillus Nidulans is an Inverted Duplication of a Low-Copy-Number Dispersed Genomic Repeat. Mol. Microbiol. 1996, 19, 565–574. [Google Scholar] [CrossRef]
- Ballantine, J.A.; Hassall, C.H.; Jones, B.D. The Biosynthesis of Phenols—XVII: Some Phenolic Metabolites of Mutant Strains of Aspergillus Regulosus. Phytochem. 1968, 7, 1529–1534. [Google Scholar] [CrossRef]
- Feng, C.; Wei, Q.; Hu, C.; Zou, Y. Biosynthesis of Diphenyl Ethers in Fungi. Org. Lett. 2019, 21, 3114–3118. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
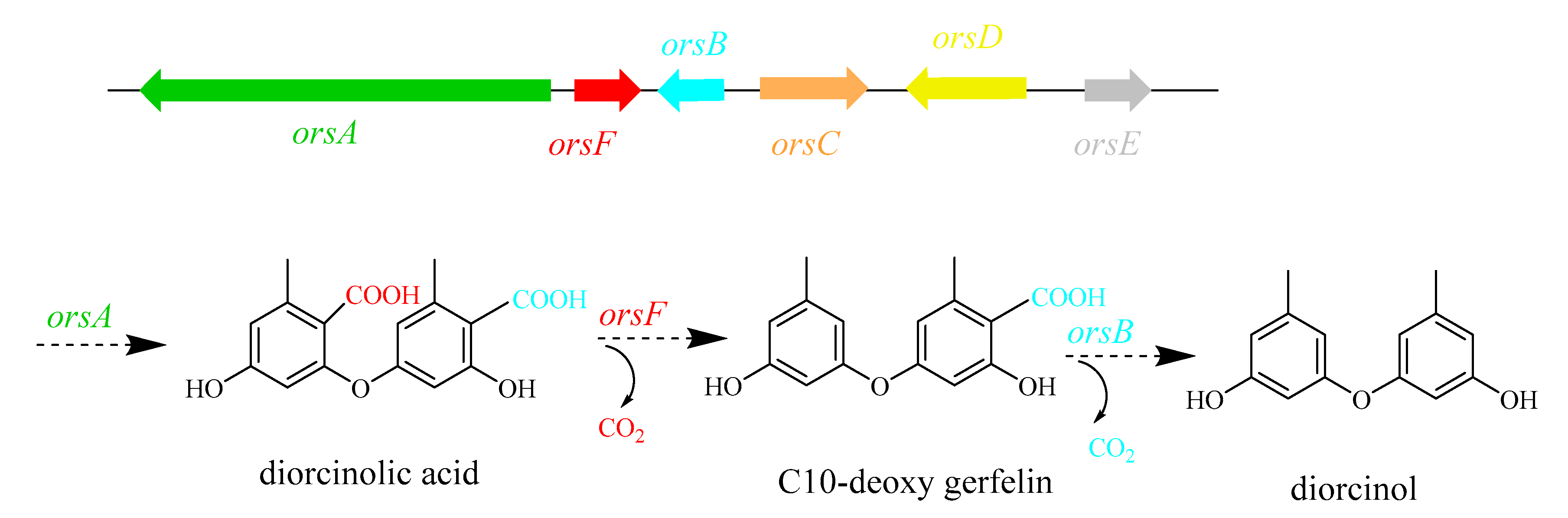
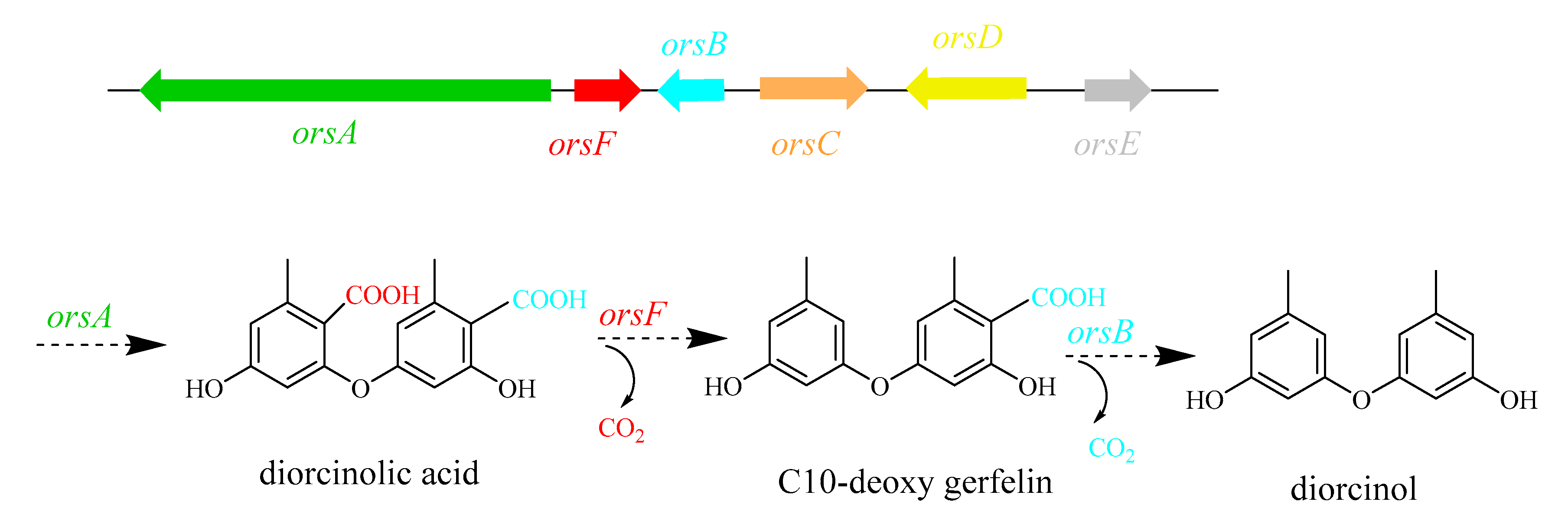
- Sanchez, J.F.; Chiang, Y.-M.; Szewczyk, E.; Davidson, A.D.; Ahuja, M.; Elizabeth Oakley, C.; Woo Bok, J.; Keller, N.; Oakley, B.R.; Wang, C.C.C. Molecular Genetic Analysis of the Orsellinic Acid/F9775 Gene Cluster of Aspergillus Nidulans. Mol. Biosyst. 2010, 6, 587–593. [Google Scholar] [CrossRef]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.J.; Wortman, J.R.; Batzoglou, S.; Lee, S.I.; Baştürkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus Nidulans and Comparative Analysis with A. Fumigatus and A. Oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Forseth, R.R.; Amaike, S.; Schwenk, D.; Affeldt, K.J.; Hoffmeister, D.; Schroeder, F.C.; Keller, N.P. Homologous NRPS-Like Gene Clusters Mediate Redundant Small-Molecule Biosynthesis in Aspergillus Flavus. Angew. Chem. Int. Ed. Engl. 2013, 52, 1590–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, S.P.; Milde, L.; Trapp, M.K.; Frisvad, J.C.; Keller, N.P.; Bok, J.W. Requirement of LaeA for Secondary Metabolism and Sclerotial Production in Aspergillus Flavus. Fungal Genet. Biol. 2008, 45, 1422–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayram, Ö.; Braus, G.H. Coordination of Secondarymetabolism and Development in Fungi: The Velvet Family of Regulatory Proteins. FEMS Microbiol. Rev. 2012, 36, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Steidl, S.; Hynes, M.J.; Brakhage, A.A. The Aspergillus Nidulans Multimeric CCAAT Binding Complex AnCF is Negatively Autoregulated via its HapB Subunit Gene. J. Mol. Biol 2001, 306, 643–653. [Google Scholar] [CrossRef]
- Ramamoorthy, V.; Dhingra, S.; Kincaid, A.; Shantappa, S.; Feng, X.; Calvo, A.M. The Putative C2H2 Transcription Factor MtfA is a Novel Regulator of Secondary Metabolism and Morphogenesis in Aspergillus Nidulans. PLoS ONE 2013, 8, e74122. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.J.; Takahashi, T.; Matsushima, K.-I.; Hara, S.; Shinohara, Y.; Maruyama, J.-I.; Kitamoto, K.; Koyama, Y. SclR, A Basic Helix-Loop-Helix Transcription Factor, Regulates Hyphal Morphology and Promotes Sclerotial Formation in Aspergillus Oryzae. Eukaryot. Cell 2011, 10, 945–955. [Google Scholar] [CrossRef] [Green Version]
- Shantappa, S.; Dhingra, S.; Hernández-Ortiz, P.; Espeso, E.A.; Calvo, A.M. Role of the Zinc Finger Transcription Factor SltA in Morphogenesis and Sterigmatocystin Biosynthesis in the Fungus Aspergillus Nidulans. PLoS ONE 2013, 8, e68492. [Google Scholar] [CrossRef] [Green Version]
- Oakley, C.E.; Ahuja, M.; Sun, W.-W.; Entwistle, R.; Akashi, T.; Yaegashi, J.; Guo, C.-J.; Cerqueira, G.C.; Russo Wortman, J.; Wang, C.C.C.; et al. Discovery of McrA, a Master Regulator of Aspergillus Secondary Metabolism. Mol. Microbiol. 2017, 103, 347–365. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.M.; Theisen, J.M.; Duran, R.M.; Grayburn, W.S.; Calvo, A.M.; Keller, N.P. Secondary Metabolism and Development is Mediated by LlmF Control of VeA Subcellular Localization in Aspergillus Nidulans. PLoS Genet. 2013, 9, e1003193. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, J.Z.; Wang, W.J.; Chen, Y.W.; Zheng, D.Q.; Di, Y.N.; Li, P.; Wang, P.M.; Li, Y.D. Genome Sequencing and Evolutionary Analysis of Marine Gut Fungus Aspergillus sp. Z5 from Ligia Oceanica. Evol. Bioinform. Online 2016, 12, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Keller, N.P. Genetic Involvement of a cAMP-Dependent Protein Kinase in a G Protein Signaling Pathway Regulating Morphological and Chemical Transitions in Aspergillus Nidulans. Genetics 2001, 157, 591. [Google Scholar] [PubMed]
- Lim, F.Y.; Sanchez, J.F.; Wang, C.C.C.; Keller, N.P. Toward Awakening Cryptic Secondary Metabolite Gene Clusters in Filamentous Fungi. Meth. Enzymol. 2012, 517, 303–324. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving Coverage, Classification and Access to Protein Sequence Annotations. Nucleic Acids Res. 2018, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Gietz, R.D.; Schiestl, R.H. High-Efficiency Yeast Transformation Using the LiAc/SS carrier DNA/PEG Method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef]
- Green, M.R. Molecular Cloning: A Laboratory Manual; Green, M.R., Sambrook, J., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012. [Google Scholar]
- Ding, W.; Zhang, H.; Mei, G. Synergistic Antitumor Activity of DHA and JQ1 in Colorectal Carcinoma. Eur. J. Pharmacol. 2020, 885, 173500. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Li, P.; Fu, X.; Li, S.; Zhang, L. Engineering TATA-Binding Protein Spt15 to Improve Ethanol Tolerance and Production in Kluyveromyces Marxianus. Biotechnol. Biofuels 2018, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Teste, M.A.; Duquenne, M.; Francois, J.M.; Parrou, J.L. Validation of Reference Genes for Quantitative Expression Analysis by Real-Time RT-PCR in Saccharomyces Cerevisiae. BMC Mol. Biol. 2009, 10, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiger, M.G.; Mach, R.L.; Mach-Aigner, A.R. An Accurate Normalization Strategy for RT-qPCR in Hypocrea Jecorina (Trichoderma reesei). J. Biotechnol. 2010, 145, 30–37. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | UV (nm) | Retention Time (min) | Width (min) | Height (mAU) | Area (mAU*s) (Average ± SE) |
|---|---|---|---|---|---|
| Aspergillus sp. Z5 | 230 | 41.58 | 0.17 | 12.5 | 152.8 ± 8.58 |
| nc10.5 | 230 | 41.61 | 0.17 | 11.7 | 151 ± 20.80 |
| OE::Az5LaeA9.8 | 230 | 41.59 | 0.17 | 52.9 | 512.3 ± 73.53 |
| OE::Az5LaeA9.10 | 230 | 41.57 | 0.18 | 35.3 | 418.9 ± 22.33 |
| OE::AnLaeA11.14 | 230 | 41.57 | 0.17 | 22.2 | 239.7 ± 15.94 |
| OE::AnLaeA11.19 | 230 | 41.60 | 0.17 | 21.8 | 340.7 ± 45.76 |
| Strains | UV (nm) | Retention Time (min) | Width (min) | Height (mAU) | Area (mAU*s) |
|---|---|---|---|---|---|
| Penicillium sp. LC1-4 | 254 | 27.66 | 0.28 | 14 | 282.4 |
| ncP10.1 | 254 | 27.68 | 0.30 | 5.1 | 109.4 |
| OE::AnLaeAPlc11.2 | 254 | 27.72 | 0.24 | 85.5 | 1376.1 |
| OE::AnLaeAPlc11.11 | 254 | 27.72 | 0.25 | 24.8 | 414.7 |
| Strain Name | Parental Strain | Genotype | Source |
|---|---|---|---|
| Aspergillus nidulans RDIT2.3 | Aspergillus nidulans | veA1 | [11] |
| Aspergillus sp. Z5 | Wild type | Wild type | [41] |
| nc10.5 | Aspergillus sp. Z5 | ama1 gpdA trpC neoR/kanR | This study |
| OE::Az5LaeA9.8 | Aspergillus sp. Z5 | ama1 gpdA::Az5LaeA::trpC neoR/kanR | This study |
| OE::Az5LaeA9.10 | Aspergillus sp. Z5 | ama1 gpdA::Az5LaeA::trpC neoR/kanR | This study |
| OE::AnLaeA11.14 | Aspergillus sp. Z5 | ama1 gpdA::AnLaeA::trpC neoR/kanR | This study |
| OE::AnLaeA11.19 | Aspergillus sp. Z5 | ama1 gpdA::AnLaeA::trpC neoR/kanR | This study |
| Penicillium sp. LC1-4 | Wild type | Wild type | |
| ncP10.1 | Penicillium sp. LC1-4 | ama1 gpdA trpC neoR/kanR | This study |
| OE::AnLaeAPlc11.2 | Penicillium sp. LC1-4 | ama1 gpdA::AnLaeA::trpC neoR/kanR | This study |
| OE::AnLaeA Plc11.11 | Penicillium sp. LC1-4 | ama1 gpdA::AnLaeA::trpC neoR/kanR | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, I.; Xie, W.-L.; Yu, Y.-C.; Sheng, H.; Xu, Y.; Wang, J.-Q.; Debnath, S.C.; Xu, J.-Z.; Zheng, D.-Q.; Ding, W.-J.; et al. Heteroexpression of Aspergillus nidulans laeA in Marine-Derived Fungi Triggers Upregulation of Secondary Metabolite Biosynthetic Genes. Mar. Drugs 2020, 18, 652. https://doi.org/10.3390/md18120652
Khan I, Xie W-L, Yu Y-C, Sheng H, Xu Y, Wang J-Q, Debnath SC, Xu J-Z, Zheng D-Q, Ding W-J, et al. Heteroexpression of Aspergillus nidulans laeA in Marine-Derived Fungi Triggers Upregulation of Secondary Metabolite Biosynthetic Genes. Marine Drugs. 2020; 18(12):652. https://doi.org/10.3390/md18120652
Chicago/Turabian StyleKhan, Ishrat, Wan-Lin Xie, Yu-Chao Yu, Huan Sheng, Yan Xu, Jia-Qi Wang, Sanjit Chandra Debnath, Jin-Zhong Xu, Dao-Qiong Zheng, Wan-Jing Ding, and et al. 2020. "Heteroexpression of Aspergillus nidulans laeA in Marine-Derived Fungi Triggers Upregulation of Secondary Metabolite Biosynthetic Genes" Marine Drugs 18, no. 12: 652. https://doi.org/10.3390/md18120652
APA StyleKhan, I., Xie, W. -L., Yu, Y. -C., Sheng, H., Xu, Y., Wang, J. -Q., Debnath, S. C., Xu, J. -Z., Zheng, D. -Q., Ding, W. -J., & Wang, P. -M. (2020). Heteroexpression of Aspergillus nidulans laeA in Marine-Derived Fungi Triggers Upregulation of Secondary Metabolite Biosynthetic Genes. Marine Drugs, 18(12), 652. https://doi.org/10.3390/md18120652