Gold Nanoparticles Using Ecklonia stolonifera Protect Human Dermal Fibroblasts from UVA-Induced Senescence through Inhibiting MMP-1 and MMP-3


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Optimization of ES-GNPs Using the ES Reductant
2.2. ES-GNPs Characterization by HR-TEM/EDS
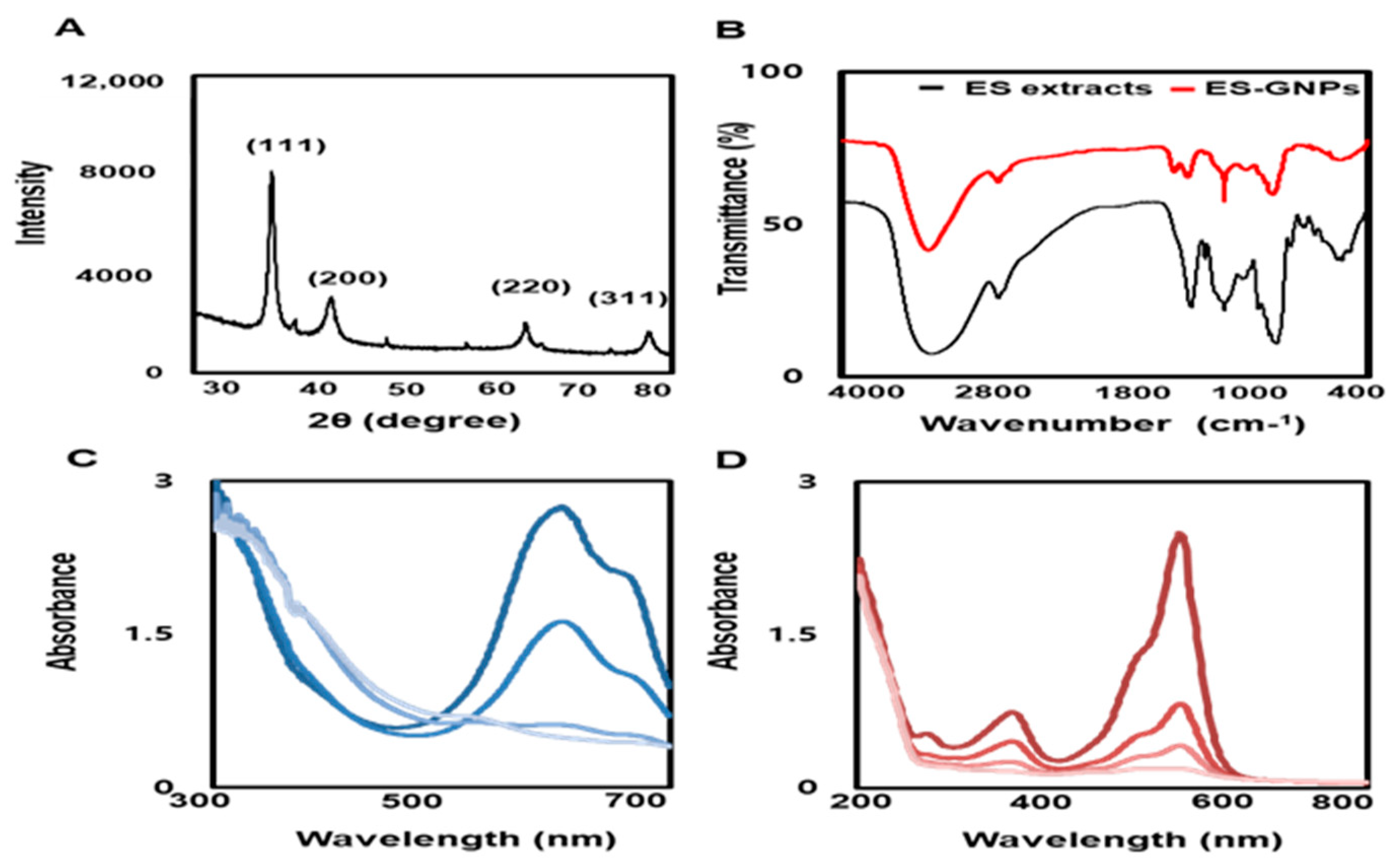
2.3. Physicochemical Characterization of ES-GNPs
2.4. ES-GNPs Ameliorate SA-β-Galactosidase Activity in UVA-Irradiated Human Dermal Fibroblasts
2.5. ES-GNPs Inhibit ROS Production and Lysosome Content in UVA-Irradiated Human Dermal Fibroblasts
2.6. ES-GNPs Inhibit G1 Arrest and Senescence-Related Proteins in UVA-Irradiated Human Dermal Fibroblasts
2.7. ES-GNPs Downregulate MMP−1/−3 mRNA, Protein Expression and Secretion in UVA-Irradiated Human Dermal Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Free Radical Scavenging Assay
4.3. Preparation of the Ecklonia stolonifera Extract
4.4. Synthesis and Physicochemical Characterization Of ES-GNPS
4.5. Photocatalytic Activities of ES-GNPS
4.6. Cell Culture and Establishment of a Cellular Model of UVA Irradiation-Induced Ssenescence
4.7. Cell Counting Kit-8 Assay
4.8. Senescence-Associated β-Galactosidase (SA-β-gal) Assay
4.9. Measurement of Intracellular ROS Production and Lysosome Content
4.10. Cell Cycle Assay
4.11. Total RNA Extraction and Quantitative Real Time PCR Analysis
4.12. MMP-1 and MMP-3 Flow Cytometry
4.13. Western Blotting Analysis
4.14. Enzyme-Linked Immunosorbent Assay (ELISA)
4.15. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nakyai, W.; Saraphanchotiwitthaya, A.; Viennet, C.; Humbert, P.; Viyoch, J. An in Vitro Model for Fibroblast Photoaging Comparing Single and Repeated UVA Irradiations. Photochem. Photobiol. 2017, 93, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Min, W.; Liu, X.; Qian, Q.; Lin, B.; Wu, D.; Wang, M.; Ahmad, I.; Yusuf, N.; Luo, D. Effects of Baicalin against UVA-Induced Photoaging in Skin Fibroblasts. Am. J. Chin. Med. 2014, 42, 709–727. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Sasaki, S.; Manabe, Y.; Hirata, T.; Sugawara, T. Preventive Effect of Dietary Astaxanthin on UVA-Induced Skin Photoaging in Hairless Mice. Plos One 2017, 12, e0171178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youn, H.J.; Kim, K.B.; Han, H.S.; An, I.S.; Ahn, K.J. 23-Hydroxytormentic Acid Protects Human Dermal Fibroblasts by Attenuating UVA-Induced Oxidative Stress. Photodermatol. Photoimmunol. Photomed. 2017, 33, 92–100. [Google Scholar] [CrossRef]
- Dunaway, S.; Odin, R.; Zhou, L.; Ji, L.; Zhang, Y.; Kadekaro, A.L. Natural Antioxidants: Multiple Mechanisms to Protect Skin from Solar Radiation. Front. Pharmacol. 2018, 9, 392. [Google Scholar] [CrossRef] [Green Version]
- Hseu, Y.C.; Korivi, M.; Lin, F.Y.; Li, M.L.; Lin, R.W.; Wu, J.J.; Yang, H.L. Trans-Cinnamic Acid Attenuates UVA-Induced Photoaging through Inhibition of AP-1 Activation and Induction of Nrf2-Mediated Antioxidant Genes in Human Skin Fibroblasts. J. Dermatol. Sci. 2018, 90, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Gendron, S.P.; Rochette, P.J. Modifications in Stromal Extracellular Matrix of Aged Corneas can be Induced by Ultraviolet A Irradiation. Aging Cell 2015, 14, 433–442. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Song, H.; He, J.; Li, G.; Zheng, Y.; Li, B. Collagen Peptides Promote Photoaging Skin Cell Repair by Activating the TGF-Beta/Smad Pathway and Depressing Collagen Degradation. Food Funct. 2019, 10, 6121–6134. [Google Scholar] [CrossRef]
- Seo, S.W.; Park, S.K.; Oh, S.J.; Shin, O.S. TLR4-Mediated Activation of the ERK Pathway Following UVA Irradiation Contributes to Increased Cytokine and MMP Expression in Senescent Human Dermal Fibroblasts. PLoS ONE 2018, 13, e0202323. [Google Scholar] [CrossRef] [Green Version]
- Philips, N.; Chalensouk-Khaosaat, J.; Gonzalez, S. Simulation of the Elastin and Fibrillin in Non-Irradiated or UVA Radiated Fibroblasts, and Direct Inhibition of Elastase or Matrix Metalloptoteinases Activity by Nicotinamide or its Derivatives. J. Cosmet. Sci. 2018, 69, 47–56. [Google Scholar]
- Chen, X.; Han, W.; Zhao, X.; Tang, W.; Wang, F. Epirubicin-Loaded Marine Carrageenan Oligosaccharide Capped Gold Nanoparticle System for pH-Triggered Anticancer Drug Release. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, X.; Gao, Y.; Yin, J.; Bai, M.; Wang, F. Green Synthesis of Gold Nanoparticles using Carrageenan Oligosaccharide and their in Vitro Antitumor Activity. Mar. Drugs 2018, 16, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Ballesteros, N.; Prado-Lopez, S.; Rodriguez-Gonzalez, J.B.; Lastra, M.; Rodriguez-Arguelles, M.C. Green Synthesis of Gold Nanoparticles using Brown Algae Cystoseira Baccata: Its Activity in Colon Cancer Cells Colloids Surf. B Biointerfaces 2017, 153, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.A.; Padil, V.V.T.; Slaveykova, V.I.; Cernik, M.; Sevcu, A. Green Synthesis of Metal and Metal Oxide Nanoparticles and their Effect on the Unicellular Alga Chlamydomonas Reinhardtii. Nanoscale Res. Lett. 2018, 13, 1–13. [Google Scholar] [CrossRef]
- Boldeiu, A.; Simion, M.; Mihalache, I.; Radoi, A.; Banu, M.; Varasteanu, P.; Nadejde, P.; Vasile, E.; Acasandrei, A.; Popescu, R.C.; et al. Comparative Analysis of Honey and Citrate Stabilized Gold Nanoparticles: In Vitro Interaction with Proteins and Toxicity Studies. J. Photochem. Photobiol. B 2019, 197, 111519. [Google Scholar] [CrossRef]
- Chahardoli, A.; Karimi, N.; Sadeghi, F.; Fattahi, A. Green Approach for Synthesis of Gold Nanoparticles from Nigella arvensis Leaf Extract and Evaluation of their Antibacterial, Antioxidant, Cytotoxicity and Catalytic Activities. Artif. Cells Nanomed. Biotechnol. 2018, 46, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Dhayalan, M.; Denison, M.I.J.; Ayyar, M.; Gandhi, N.N.; Krishnan, K.; Abdulhadi, B. Biogenic Synthesis, Characterization of Gold and Silver Nanoparticles from Coleus forskohlii and their Clinical Importance. J. Photochem. Photobiol. B 2018, 183, 251–257. [Google Scholar] [CrossRef]
- Park, S.Y.; Yi, E.H.; Kim, Y.; Park, G. Anti-Neuroinflammatory Effects of Ephedra Sinica Stapf Extract-Capped Gold Nanoparticles in Microglia. Int. J. Nanomed. 2019, 14, 2861–2877. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, M.T.; Bangoura, I.; Kang, J.Y.; Cho, J.Y.; Joo, J.; Choi, Y.S.; Hwang, D.S.; Hong, Y.K. Comparison of Ecklonia cava, Ecklonia stolonifera and Eisenia bicyclis for Phlorotannin Extraction. J. Environ. Biol. 2014, 35, 713–719. [Google Scholar]
- Manandhar, B.; Wagle, A.; Seong, S.H.; Paudel, P.; Kim, H.R.; Jung, H.A.; Choi, J.S. Phlorotannins with Potential Anti-Tyrosinase and Antioxidant Activity Isolated from the Marine Seaweed Ecklonia stolonifera. Antioxidants 2019, 8, 240. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.H.; Choi, J.S.; Nam, T.J. Fucosterol from an Edible Brown Alga Ecklonia stolonifera Prevents Soluble Amyloid Beta-Induced Cognitive Dysfunction in Aging Rats. Mar. Drugs 2018, 16, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, J.H.; Kim, J.; Choung, S.Y. Hepaprotective Effect of Standardized Ecklonia stolonifera Formulation on CCl4-Induced Liver Injury in Sprague-Dawley Rats. Biomol. Ther. 2018, 26, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, T.S.; Kim, S.K.; Ryu, B.; Ngo, D.H.; Yoon, N.Y.; Bach, L.G.; Hang, N.T.N.; Ngo, D.N. The Suppressive Activity of Fucofuroeckol-A Derived from Brown Algal Ecklonia stolonifera Okamura on UVB-Induced Mast Cell Degranulation. Mar. Drugs 2018, 16, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Jung, H.A.; Kang, M.J.; Choi, J.S.; Kim, G.D. Fucosterol, Isolated from Ecklonia stolonifera, Inhibits Adipogenesis through Modulation of FoxO1 Pathway in 3T3-L1 Adipocytes. J. Pharm. Pharmacol. 2017, 69, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Ali, M.Y.; Choi, R.J.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Kinetics and Molecular Docking Studies of Fucosterol and Fucoxanthin, BACE1 Inhibitors from Brown Algae Undaria pinnatifida and Ecklonia stolonifera. Food Chem. Toxicol. 2016, 89, 104–111. [Google Scholar] [CrossRef]
- Choi, J.S.; Han, Y.R.; Byeon, J.S.; Choung, S.Y.; Sohn, H.S.; Jung, H.A. Protective Effect of Fucosterol Isolated from the Edible Brown Algae, Ecklonia stolonifera and Eisenia bicyclis, on Tert-Butyl Hydroperoxide- and Tacrine-Induced HepG2 Cell Injury. J. Pharm. Pharmacol. 2015, 67, 1170–1178. [Google Scholar] [CrossRef]
- Keijok, W.J.; Pereira, R.H.A.; Alvarez, L.A.C.; Prado, A.R.; da Silva, A.R.; Ribeiro, J.; de Oliveira, J.P.; Guimaraes, M.C.C. Controlled Biosynthesis of Gold Nanoparticles with Coffea arabica using Factorial Design. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Qin, H.; Zhang, G.; Zhang, L. GSK126 (EZH2 Inhibitor) Interferes with Ultraviolet A Radiation-Induced Photoaging of Human Skin Fibroblast Cells. Exp. Ther. Med. 2018, 15, 3439–3448. [Google Scholar] [CrossRef]
- Kim, K.J.; Xuan, S.H.; Park, S.N. Licoricidin, an Isoflavonoid Isolated from Glycyrrhiza Uralensis Fisher, Prevents UVA-Induced Photoaging of Human Dermal Fibroblasts. Int. J. Cosmet. Sci. 2017, 39, 133–140. [Google Scholar] [CrossRef]
- Vijayan, R.; Joseph, S.; Mathew, B. Anticancer, Antimicrobial, Antioxidant, and Catalytic Activities of Green-Synthesized Silver and Gold Nanoparticles using Bauhinia purpurea Leaf Extract. Bioprocess. Biosyst. Eng. 2019, 42, 305–319. [Google Scholar] [CrossRef]
- Young Park, S.; Jin Kim, Y.; Park, G.; Kim, H.H. Neuroprotective Effect of Dictyopteris Divaricata Extract-Capped Gold Nanoparticles against Oxygen and Glucose Deprivation/Reoxygenation. Colloids Surf. B Biointerfaces 2019, 179, 421–428. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jun, E.-S.; Kim, Y.J.; Kim, H.-H.; Park, S.Y. Gold Nanoparticles Using Ecklonia stolonifera Protect Human Dermal Fibroblasts from UVA-Induced Senescence through Inhibiting MMP-1 and MMP-3. Mar. Drugs 2020, 18, 433. https://doi.org/10.3390/md18090433
Jun E-S, Kim YJ, Kim H-H, Park SY. Gold Nanoparticles Using Ecklonia stolonifera Protect Human Dermal Fibroblasts from UVA-Induced Senescence through Inhibiting MMP-1 and MMP-3. Marine Drugs. 2020; 18(9):433. https://doi.org/10.3390/md18090433
Chicago/Turabian StyleJun, Eun-Sook, Yeong Jin Kim, Hyung-Hoi Kim, and Sun Young Park. 2020. "Gold Nanoparticles Using Ecklonia stolonifera Protect Human Dermal Fibroblasts from UVA-Induced Senescence through Inhibiting MMP-1 and MMP-3" Marine Drugs 18, no. 9: 433. https://doi.org/10.3390/md18090433
APA StyleJun, E.-S., Kim, Y. J., Kim, H.-H., & Park, S. Y. (2020). Gold Nanoparticles Using Ecklonia stolonifera Protect Human Dermal Fibroblasts from UVA-Induced Senescence through Inhibiting MMP-1 and MMP-3. Marine Drugs, 18(9), 433. https://doi.org/10.3390/md18090433