Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge


Abstract
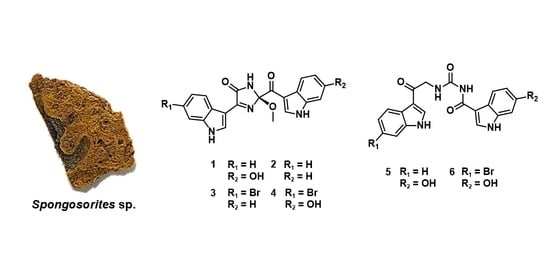
1. Introduction
2. Results and Discussion
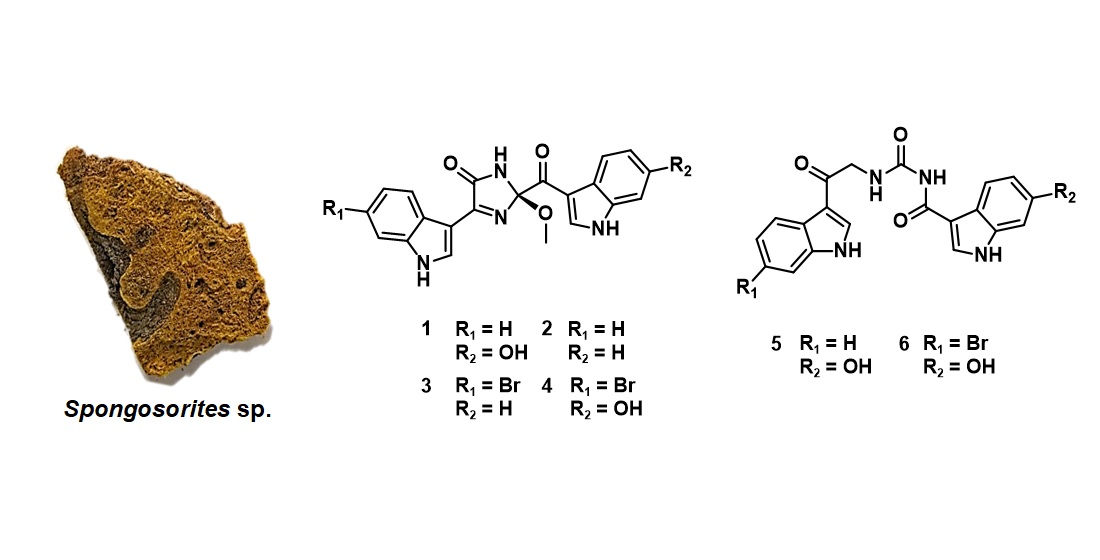
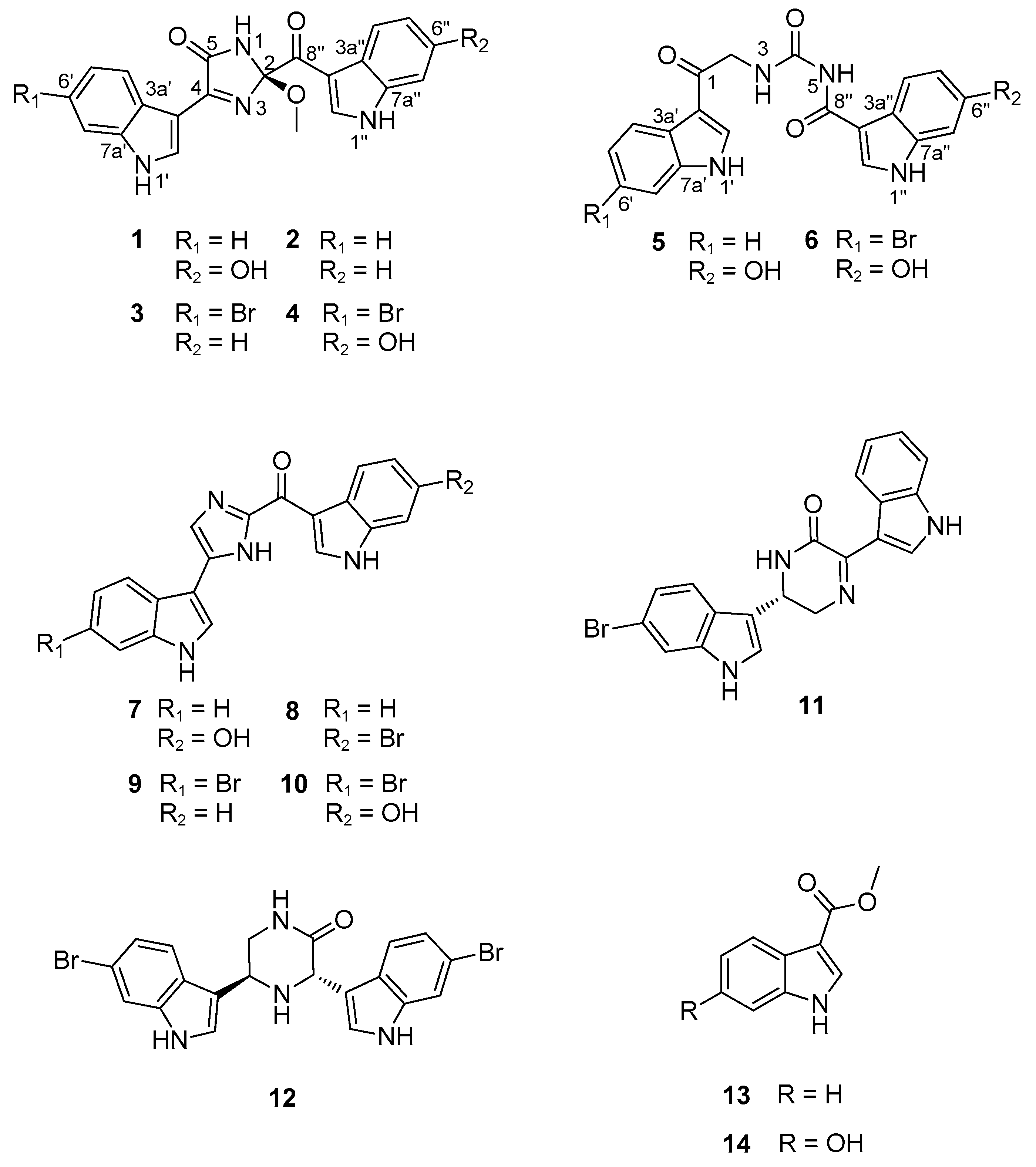
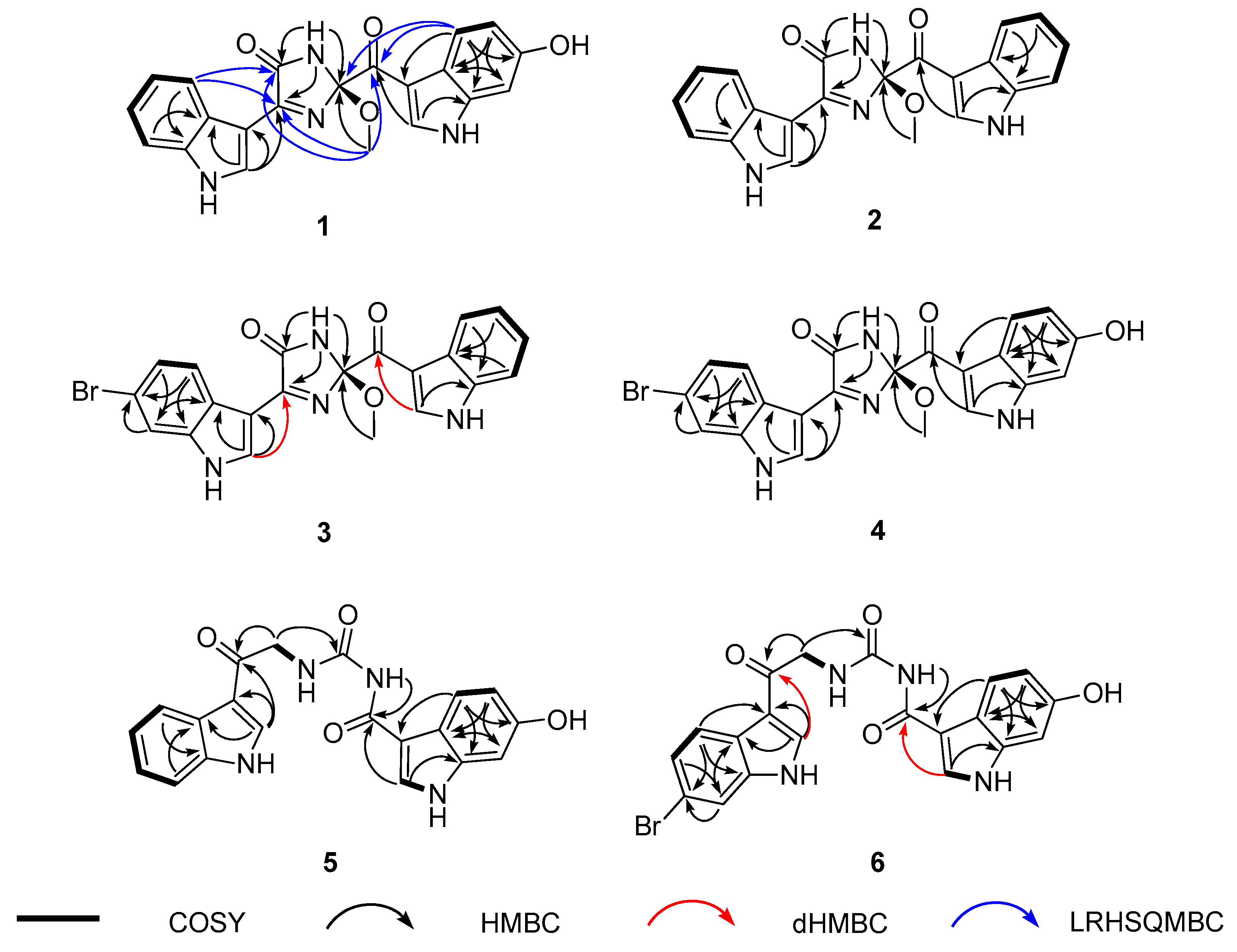
2.1. Structure Elucidation
2.2. Biological Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. ECD Calculations
3.5. SrtA Inhibition Assay
3.6. Antibacterial, Antifungal, and Cytotoxic Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cordell, G.A.; Quinn-Beattie, M.L.; Farnsworth, N.R. The potential of alkaloids in drug discovery. Phytother. Res. 2001, 15, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Kochanowska-Karamyan, A.J.; Hamann, M.T. Marine Indole Alkaloids: Potential New Drug Leads for the Control of Depression and Anxiety. Chem. Rev. 2010, 110, 4489–4497. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.P.; Or, T.C.T.; Ip, N.Y.; Cho, T.; Or, T. Plant alkaloids as drug leads for Alzheimer’s disease. Neurochem. Int. 2015, 89, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Huang, G. The synthesis and biological activity of marine alkaloid derivatives and analogues. RSC Adv. 2020, 10, 31909–31935. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175. [Google Scholar] [CrossRef]
- Schmidt, E.W.; Harper, M.K.; Faulkner, D.J. Makaluvamines H-M and Damirone C from the Pohnpeian Sponge Zyzzya fuliginosa. J. Nat. Prod. 1995, 58, 1861–1867. [Google Scholar] [CrossRef]
- Sakai, R.; Higa, T.; Jefford, C.W.; Bernardinelli, G. Manzamine A, a novel antitumor alkaloid from a sponge. J. Am. Chem. Soc. 1986, 108, 6404–6405. [Google Scholar] [CrossRef]
- Kochanowska-Karamyan, A.J.; Araujo, H.C.; Zhang, X.; El-Alfy, A.; Carvalho, P.; Avery, M.A.; Holmbo, S.D.; Magolan, J.; Hamann, M.T. Isolation and Synthesis of Veranamine, an Antidepressant Lead from the Marine Sponge Verongula rigida. J. Nat. Prod. 2020, 83, 1092–1098. [Google Scholar] [CrossRef]
- Ma, Y.; Yakushijin, K.; Miyake, F.; Horne, D. A concise synthesis of indolic enamides: Coscinamide A, coscinamide B, and igzamide. Tetrahedron Lett. 2009, 50, 4343–4345. [Google Scholar] [CrossRef]
- Guella, G.; Mancini, I.; Zibrowius, H.; Pietra, F. Aplysinopsin-type alkaloids fromDendrophyllia sp., a scleractinian coral of the family dendrophylliidae of the philippines, facile photochemical (Z/E) photoisomerization and thermal reversal. Helvetica Chim. Acta 1989, 72, 1444–1450. [Google Scholar] [CrossRef]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef]
- Ang, K.K.H.; Holmes, M.J.; Higa, T.; Hamann, M.T.; Kara, U.A.K. In Vivo Antimalarial Activity of the Beta-Carboline Alkaloid Manzamine A. Antimicrob. Agents Chemother. 2000, 44, 1645–1649. [Google Scholar] [CrossRef] [PubMed]
- Yamanokuchi, R.; Imada, K.; Miyazaki, M.; Kato, H.; Watanabe, T.; Fujimuro, M.; Saeki, Y.; Yoshinaga, S.; Terasawa, H.; Iwasaki, N.; et al. Hyrtioreticulins A–E, indole alkaloids inhibiting the ubiquitin-activating enzyme, from the marine sponge Hyrtios reticulatus. Bioorganic Med. Chem. 2012, 20, 4437–4442. [Google Scholar] [CrossRef] [PubMed]
- Shinkre, B.A.; Raisch, K.P.; Fan, L.; Velu, S.E. Analogs of the marine alkaloid makaluvamines: Synthesis, topoisomerase II inhibition, and anticancer activity. Bioorganic Med. Chem. Lett. 2007, 17, 2890–2893. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Seo, Y.; Cho, K.W.; Rho, J.-R.; Sim, C.J. New Bis(Indole) Alkaloids of the Topsentin Class from the SpongeSpongosorites genitrix. J. Nat. Prod. 1999, 62, 647–649. [Google Scholar] [CrossRef]
- Guinchard, X.; Vallee, Y.; Denis, J.-N. Total Synthesis of Marine Sponge Bis(indole) Alkaloids of the Topsentin Class. J. Org. Chem. 2007, 72, 3972–3975. [Google Scholar] [CrossRef]
- Oh, K.-B.; Mar, W.; Kim, S.; Kim, J.-Y.; Oh, M.-N.; Kim, J.-G.; Shin, D.; Sim, C.J.; Shin, J. Bis(indole) alkaloids as sortase A inhibitors from the sponge Spongosorites sp. Bioorganic Med. Chem. Lett. 2005, 15, 4927–4931. [Google Scholar] [CrossRef]
- Keyzers, R.A.; Davies-Coleman, M.T. Anti-inflammatory metabolites from marine sponges. Chem. Soc. Rev. 2005, 34, 355–365. [Google Scholar] [CrossRef]
- Oh, K.-B.; Mar, W.; Kim, S.; Kim, J.-Y.; Lee, T.-H.; Kim, J.-G.; Shin, D.; Sim, C.J.; Shin, J. Antimicrobial Activity and Cytotoxicity of Bis(indole) Alkaloids from the Sponge Spongosorites sp. Biol. Pharm. Bull. 2006, 29, 570–573. [Google Scholar] [CrossRef]
- Yang, C.-G.; Huang, H.; Jiang, B. Progress in Studies of Novel Marine Bis(indole) Alkaloids. Curr. Org. Chem. 2004, 8, 1691–1720. [Google Scholar] [CrossRef]
- Hwang, J.; Kim, D.; Park, J.S.; Park, H.J.; Shin, J.; Lee, S.K. Park Photoprotective Activity of Topsentin, A Bis(Indole) Alkaloid from the Marine Sponge Spongosorites genitrix, by Regulation of COX-2 and Mir-4485 Expression in UVB-Irradiated Human Keratinocyte Cells. Mar. Drugs 2020, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Pandita, N.S.; Pal, A.K. LC-MS/MS as a tool for identification of bioactive compounds in marine sponge Spongosorites halichondriodes (Dendy 1905). Toxicon 2012, 60, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.K.; Sarpong, R.; Stoltz, B.M. The First Total Synthesis of Dragmacidin D. J. Am. Chem. Soc. 2002, 124, 13179–13184. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.P.; McCarthy, P.J.; Kelly-Borges, M. Hamacanthins A and B, New Antifungal Bis Indole Alkaloids from the Deep-Water Marine Sponge, Hamacantha Sp. J. Nat. Prod. 1994, 57, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Tsuda, M.; Fromont, J.; Kobayashi, J. Hyrtinadine A, a Bis-indole Alkaloid from a Marine Sponge. J. Nat. Prod. 2007, 70, 423–424. [Google Scholar] [CrossRef]
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.-H.; Cirrincione, G. Synthesis and antitumor activity of 2,5-bis(3′-indolyl)-furans and 3,5-bis(3′-indolyl)-isoxazoles, nortopsentin analogues. Bioorganic Med. Chem. 2010, 18, 4524–4529. [Google Scholar] [CrossRef]
- Sato, H.; Tsuda, M.; Watanabe, K.; Kobayashi, J. Rhopaladins A ∼ D, new indole alkaloids from marine tunicate Rhopalaea sp. Tetrahedron 1998, 54, 8687–8690. [Google Scholar] [CrossRef]
- Jennings, L.K.; Khan, N.M.D.; Kaur, N.; Rodrigues, D.; Morrow, C.; Boyd, A.; Thomas, O.P. Brominated Bisindole Alkaloids from the Celtic Sea Sponge Spongosorites calcicola. Molecules 2019, 24, 3890. [Google Scholar] [CrossRef]
- Williamson, R.T.; Buevich, A.V.; Martin, G.E.; Parella, T. LR-HSQMBC: A Sensitive NMR Technique To Probe Very Long-Range Heteronuclear Coupling Pathways. J. Org. Chem. 2014, 79, 3887–3894. [Google Scholar] [CrossRef]
- Pecul, M.; Ruud, K.; Rizzo, A.; Helgaker, T. Conformational Effects on the Optical Rotation of Alanine and Proline. J. Phys. Chem. A 2004, 108, 4269–4276. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Vaccaro, P.H.; Cheeseman, J.R. Conformational Effects on Optical Rotation. 3-Substituted 1-Butenes. J. Am. Chem. Soc. 2003, 125, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Grkovic, T.; Ding, Y.; Li, X.-C.; Webb, V.L.; Ferreira, D.; Copp, B.R. Enantiomeric Discorhabdin Alkaloids and Establishment of Their Absolute Configurations Using Theoretical Calculations of Electronic Circular Dichroism Spectra. J. Org. Chem. 2008, 73, 9133–9136. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.-E.; Na, Z.; Jung, M.; Lee, H.-S.; Sim, C.J.; Nahm, K.; Oh, K.-B.; Shin, J. Discorhabdins from the Korean Marine SpongeSceptrellasp. J. Nat. Prod. 2010, 73, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Cason, J.; Koch, C.W.; Correia, J.S. Structures of herqueinone, isoherqueinone and norherqueinone. J. Org. Chem. 1970, 35, 179–186. [Google Scholar] [CrossRef]
- Park, S.C.; Julianti, E.; Ahn, S.; Kim, D.; Lee, S.K.; Noh, M.; Oh, D.-C.; Oh, K.-B.; Shin, A.J.; Shin, J. Phenalenones from a Marine-Derived Fungus Penicillium Sp. Mar. Drugs 2019, 17, 176. [Google Scholar] [CrossRef]
- Bae, J.; Cho, E.; Park, J.S.; Won, T.H.; Seo, S.-Y.; Oh, D.-C.; Oh, K.-B.; Shin, J. Isocadiolides A–H: Polybrominated Aromatics from a Synoicum sp. Ascidian. J. Nat. Prod. 2020, 83, 429–437. [Google Scholar] [CrossRef]
- Zhang, Z.-W.; Xue, H.; Li, H.; Kang, H.; Feng, J.; Lin, A.; Liu, S. Collective Synthesis of 3-Acylindoles, Indole-3-carboxylic Esters, Indole-3-sulfinic Acids, and 3-(Methylsulfonyl)indoles from Free (N–H) Indoles via Common N-Indolyl Triethylborate. Org. Lett. 2016, 18, 3918–3921. [Google Scholar] [CrossRef]
- Casapullo, A.; Bifulco, G.; Bruno, I.; Riccio, R. New Bisindole Alkaloids of the Topsentin and Hamacanthin Classes from the Mediterranean Marine SpongeRhaphisia lacazei. J. Nat. Prod. 2000, 63, 447–451. [Google Scholar] [CrossRef]
- Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C.-O.; Cho, H.Y.; Jung, J.H. Bisindole Alkaloids of the Topsentin and Hamacanthin Classes from a Marine SpongeSpongosoritessp. J. Nat. Prod. 2007, 70, 2–8. [Google Scholar] [CrossRef]
- Bao, B.; Zhang, P.; Lee, Y.; Hong, J.; Lee, C.-O.; Jung, J.H. Monoindole Alkaloids from a Marine Sponge Spongosorites sp. Mar. Drugs 2007, 5, 31–39. [Google Scholar] [CrossRef]
- Kai, S.; Suzuki, M. ChemInform Abstract: Dye-Sensitized Photooxidation of 2,4-Disubstituted Imidazoles: The Formation of Isomeric Imidazolinones. ChemInform 2010, 27, 1185–1188. [Google Scholar] [CrossRef]
- Maresso, A.W.; Schneewind, O. Sortase as a Target of Anti-Infective Therapy. Pharmacol. Rev. 2008, 60, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Totsika, M.; Schillaci, D. Sortase A: An ideal target for anti-virulence drug development. Microb. Pathog. 2014, 77, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-B.; Kim, S.-H.; Lee, J.; Cho, W.-J.; Lee, T.; Kim, S. Discovery of Diarylacrylonitriles as a Novel Series of Small Molecule Sortase A Inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C.; Chung, B.; Lee, J.; Cho, E.; Hwang, J.-Y.; Oh, D.-C.; Shin, J.; Oh, K.-B. Sortase A-Inhibitory Metabolites from a Marine-Derived Fungus Aspergillus sp. Mar. Drugs 2020, 18, 359. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Ton-That, H.; Su, K.; Schneewind, O. An iron-regulated sortase anchors a class of surface protein during Staphylococcus aureus pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 2293–2298. [Google Scholar] [CrossRef]
- Park, J.-S.; Chung, B.; Lee, W.-H.; Lee, J.; Suh, Y.; Oh, D.-C.; Oh, K.-B.; Shin, J. Sortase A-Inhibitory Coumarins from the Folk Medicinal Plant Poncirus trifoliata. J. Nat. Prod. 2020, 83, 3004–3011. [Google Scholar] [CrossRef]
- Cho, E.; Kwon, O.-S.; Chung, B.; Lee, J.; Sun, J.; Shin, J.; Oh, K.-B. Antibacterial Activity of Chromomycins from a Marine-Derived Streptomyces microflavus. Mar. Drugs 2020, 18, 522. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
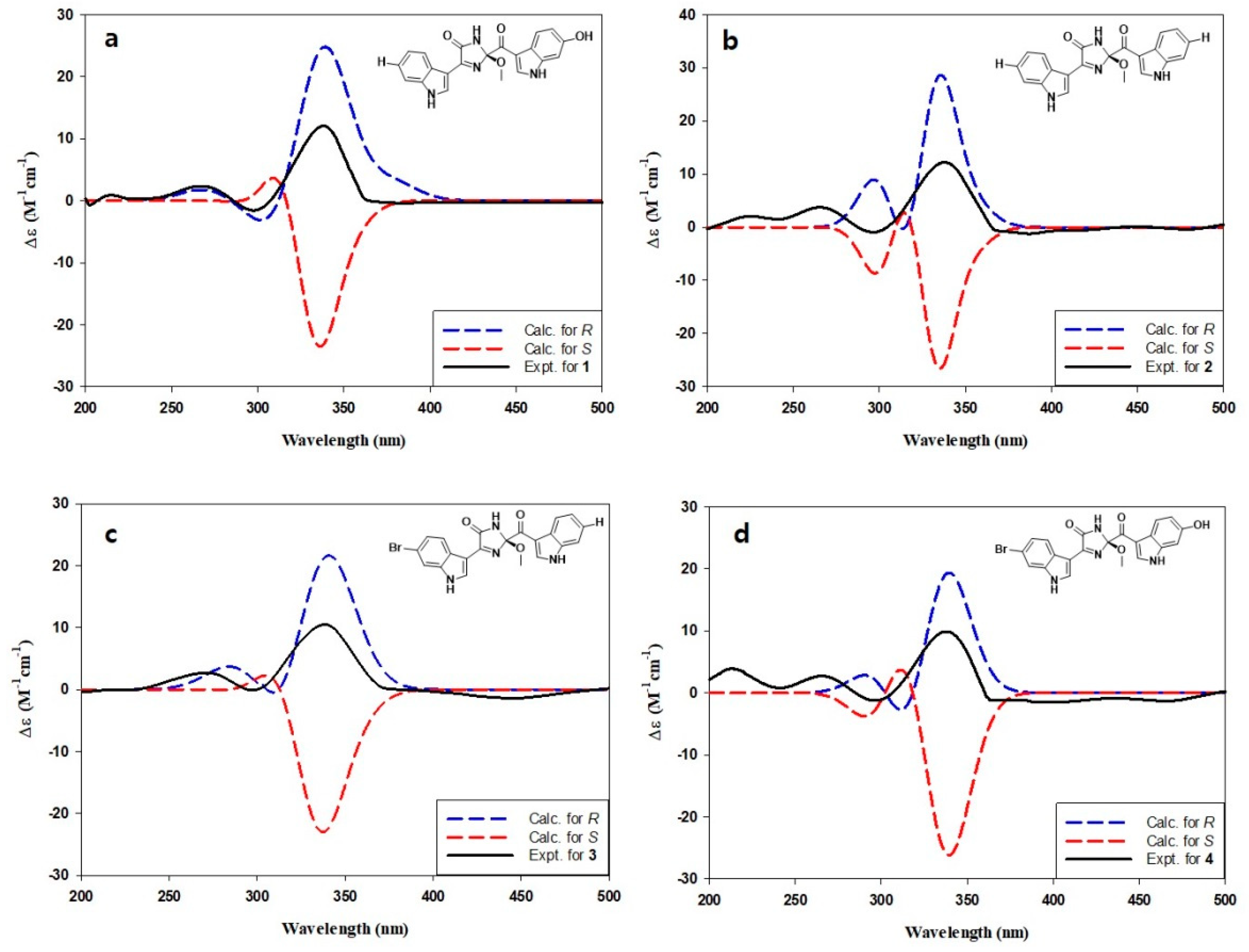{kind=link}
| No. | 1 b | 2 b | 3 b | 4 b | ||||
|---|---|---|---|---|---|---|---|---|
| ppm, Type | δ, mult (J in Hz) | ppm, Type | δ, mult (J in Hz) | ppm, Type | δ, mult (J in Hz) | ppm, Type | δ, mult (J in Hz) | |
| 1 | 10.10, s | 10.20, s | 10.20, s | 10.10, s | ||||
| 2 | 106.9, C | 106.9, C | 107.0, C | 107.0, C | ||||
| 4 | 160.1, C | 161.1, C | 161.1, C | 160.9, C | ||||
| 5 | 166.4, C | 166.4, C | 166.1, C | 166.2, C | ||||
| 1′ | 12.00, s | ND c | 12.10, s | 11.90, s | ||||
| 2′ | 132.9, CH | 8.67, s | 133.0, CH | 8.68, s | 133.9, CH | 8.69, d (3.0) | 132.9, CH | 8.67, s |
| 3′ | 105.9, C | 105.9, C | 106.0, C | 106.0, C | ||||
| 3a′ | 125.6, C | 125.6, C | 124.6, C | 124.4, C | ||||
| 4′ | 121.5, CH | 8.18, d (7.8) | 121.5, CH | 8.18, d (7.8) | 123.1, CH | 8.17, d (7.8) | 123.1, CH | 8.09, d (8.2) |
| 5′ | 121.5, CH | 7.16, dd (8.0, 7.8) | 121.6, CH | 7.16, dd (7.8, 7.0) | 124.4, CH | 7.30, dd (7.8, 1.5) | 124.6, CH | 7.30, dd (8.2, 1.9) |
| 6′ | 123.1, CH | 7.23, dd (8.0, 7.4) | 123.1, CH | 7.23, dd (8.0, 7.0) | 115.7, C | 115.1, C | ||
| 7′ | 112.2, CH | 7.51, d (8.0) | 112.4, CH | 7.51, d (8.0) | 115.1, CH | 7.72, d (1.5) | 115.7, CH | 7.72, d (1.9) |
| 7a′ | 136.4, C | 136.5, C | 137.4, C | 137.4, C | ||||
| 1′′ | 11.80, s | ND c | 12.20, s | ND c | ||||
| 2′′ | 136.4, CH | 8.55, s | 137.7, CH | 8.74, s | 137.7, CH | 8.73, d (3.0) | 123.1, CH | 8.53, s |
| 3′′ | 112.3, C | 112.1, C | 112.1, C | 112.1, C | ||||
| 3a′′ | 119.5, C | 126.7, C | 126.7, C | 119.5, C | ||||
| 4′′ | 121.7, CH | 7.92, d (8.6) | 121.2, CH | 8.14, d (7.8) | 121.2, CH | 8.09, d (8.2) | 121.7, CH | 7.91, d (8.4) |
| 5′′ | 112.2, CH | 6.70, dd (8.6, 2.0) | 122.2, CH | 7.19, dd (7.8, 7.6) | 122.3, CH | 7.22, dd (8.2, 7.7) | 112.7, CH | 6.69, dd (8.4, 2.1) |
| 6′′ | 154.2, C | 123.0, CH | 7.24, dd (8.0, 7.6) | 123.0, CH | 7.22, dd (7.7, 7.6) | 154.3, C | ||
| 7′′ | 97.5, CH | 6.87, d (2.0) | 112.4, CH | 7.55, d (8.0) | 112.4, CH | 7.54, d (7.6) | 97.5, CH | 6.88, d (2.1) |
| 7a′′ | 137.1, C | 136.0, C | 135.9, C | 137.1, C | ||||
| 8′′ | 186.2, C | 186.5, C | 186.3, C | 185.9, C | ||||
| 2-OMe | 49.3, CH3 | 3.28, s | 49.3, CH3 | 3.30, s | 49.3, CH3 | 3.30, s | 49.4, CH3 | 3.28, s |
| No. | 5 b | 6 b | ||
|---|---|---|---|---|
| ppm, Type | δ, mult (J in Hz) | ppm, Type | δ, mult (J in Hz) | |
| 1 | 189.7, C | 189.7, C | ||
| 2 | 46.5, CH2 | 4.71, d (5.2) | 46.6, CH2 | 4.74, d (5.2) |
| 3 | 9.35, t (5.2) | 9.33, t (5.2) | ||
| 4 | 154.5, C | 154.5, C | ||
| 5 | 10.20, s | 10.20, s | ||
| 1′ | 12.00, s | NDc | ||
| 2′ | 133.7, CH | 8.47, s | 134.6, CH | 8.50, s |
| 3′ | 113.8, C | 113.9, C | ||
| 3a′ | 125.4, C | 124.4, C | ||
| 4′ | 121.1, CH | 8.18, d (7.8) | 122.8, CH | 8.11, d (8.4) |
| 5′ | 121.9, CH | 7.22, dd (7.8, 7.0) | 128.4, CH | 7.36, dd (8.4, 1.6) |
| 6′ | 122.9, CH | 7.23, dd (8.0, 7.0) | 114.9, C | |
| 7′ | 112.2, CH | 7.49, d (8.0) | 115.5, CH | 7.69, d (1.6) |
| 7a′ | 136.5, C | 137.4, C | ||
| 1′′ | 11.50, s | 11.60, s | ||
| 2′′ | 129.3, CH | 8.34, d (2.2) | 129.2, CH | 8.33, s |
| 3′′ | 108.4, C | 108.4, C | ||
| 3a′′ | 119.5, C | 119.4, C | ||
| 4′′ | 121.5, CH | 7.96, d (8.5) | 121.5, CH | 7.95, d (8.6) |
| 5′′ | 111.8, CH | 6.70, dd (8.5, 2.0) | 111.8, CH | 6.74, dd (8.6, 2.0) |
| 6′′ | 154.0, C | 154.0, C | ||
| 7′′ | 97.1, CH | 6.80, d (2.0) | 97.1, CH | 6.84, d (2.0) |
| 7a′′ | 137.6, C | 137.6, C | ||
| 8′′ | 165.5, C | 165.5, C | ||
| Antibacterial Activity MIC (μg/mL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gram (+) Positive | Gram (−) Negative | IC50 (μM) | |||||||
| No. | S. aureus | Enterococcus faecalis | Enterococcus faecium | Klebsiella pneumonia | Salmonella enterica | E. coli | Srt A | A549 | K562 |
| 1 | >128 | >128 | >128 | >128 | >128 | >128 | >329.8 | 77.3 | 24.2 |
| 2 | 64 | >128 | >128 | 128 | 128 | >128 | 62.7 | 55.7 | 28.5 |
| 3 | 32 | 128 | >128 | >128 | 64 | >128 | 43.9 | 61.2 | 37.7 |
| 4 | 16 | 128 | 128 | >128 | 64 | >128 | >274.7 | 70.9 | 54.2 |
| 5 | >128 | >128 | >128 | >128 | >128 | >128 | 79.4 | >100 | 92.8 |
| 6 | 64 | >128 | >128 | >128 | 128 | >128 | 52.4 | >100 | >100 |
| 7 | 32 | 64 | 64 | 64 | 64 | >128 | >374.1 | 25.3 | 15.3 |
| 8 | 32 | >128 | >128 | >128 | >128 | >128 | 41.3 | 22.7 | 12.5 |
| 9 | 32 | >128 | >128 | >128 | >128 | >128 | 34.0 | 29.5 | 14.4 |
| 10 | 8 | 32 | 16 | 64 | 16 | >128 | 53.2 | 38.9 | 43.2 |
| 11 | 8 | 32 | 32 | 8 | 16 | >128 | 47.5 | 39.6 | >100 |
| 12 | 8 | 16 | >128 | 8 | 8 | >128 | >263.4 | 34.3 | 9.7 |
| 13 | 128 | >128 | >128 | >128 | >128 | >128 | 164.4 | 59.2 | >100 |
| 14 | 128 | >128 | >128 | >128 | >128 | >128 | 191.1 | 66.8 | >100 |
| ampicillin | 0.13 | 0.5 | 1 | ND a | 0.25 | 8 | |||
| tetracycline | ND a | ND a | ND a | 0.25 | ND a | ND a | |||
| triphasiol | 37.9 | ||||||||
| berberine chloride | 87.4 | ||||||||
| doxorubicin | 0.64 | 0.92 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.S.; Cho, E.; Hwang, J.-Y.; Park, S.C.; Chung, B.; Kwon, O.-S.; Sim, C.J.; Oh, D.-C.; Oh, K.-B.; Shin, J. Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge. Mar. Drugs 2021, 19, 3. https://doi.org/10.3390/md19010003
Park JS, Cho E, Hwang J-Y, Park SC, Chung B, Kwon O-S, Sim CJ, Oh D-C, Oh K-B, Shin J. Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge. Marine Drugs. 2021; 19(1):3. https://doi.org/10.3390/md19010003
Chicago/Turabian StylePark, Jae Sung, Eunji Cho, Ji-Yeon Hwang, Sung Chul Park, Beomkoo Chung, Oh-Seok Kwon, Chung J. Sim, Dong-Chan Oh, Ki-Bong Oh, and Jongheon Shin. 2021. "Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge" Marine Drugs 19, no. 1: 3. https://doi.org/10.3390/md19010003
APA StylePark, J. S., Cho, E., Hwang, J.-Y., Park, S. C., Chung, B., Kwon, O.-S., Sim, C. J., Oh, D.-C., Oh, K.-B., & Shin, J. (2021). Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge. Marine Drugs, 19(1), 3. https://doi.org/10.3390/md19010003