
Investigation of Marine-Derived Natural Products as Raf Kinase Inhibitory Protein (RKIP)-Binding Ligands




Abstract
:1. Introduction
2. Results
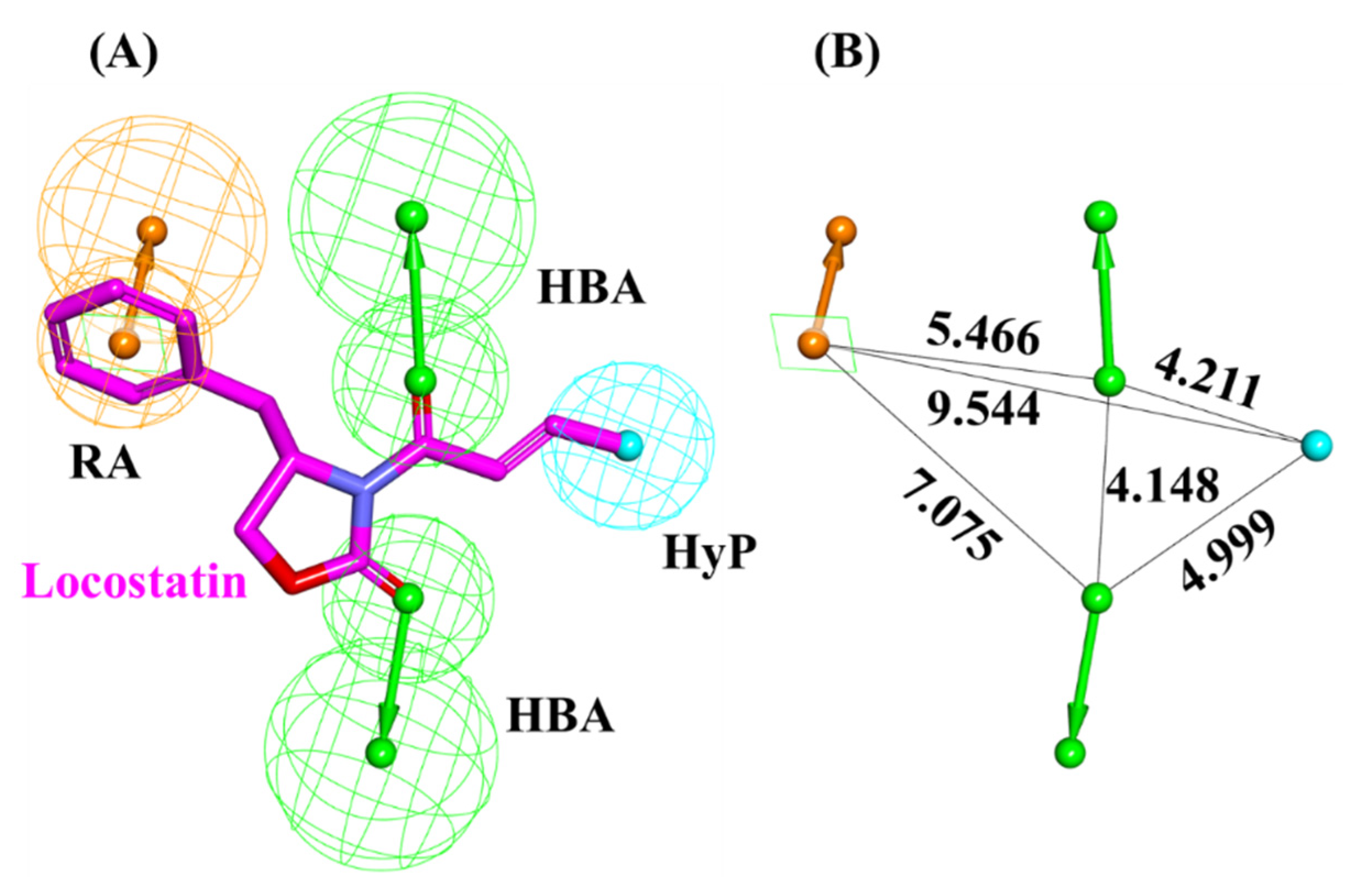
2.1. Generated Auto-Pharmacophore Model
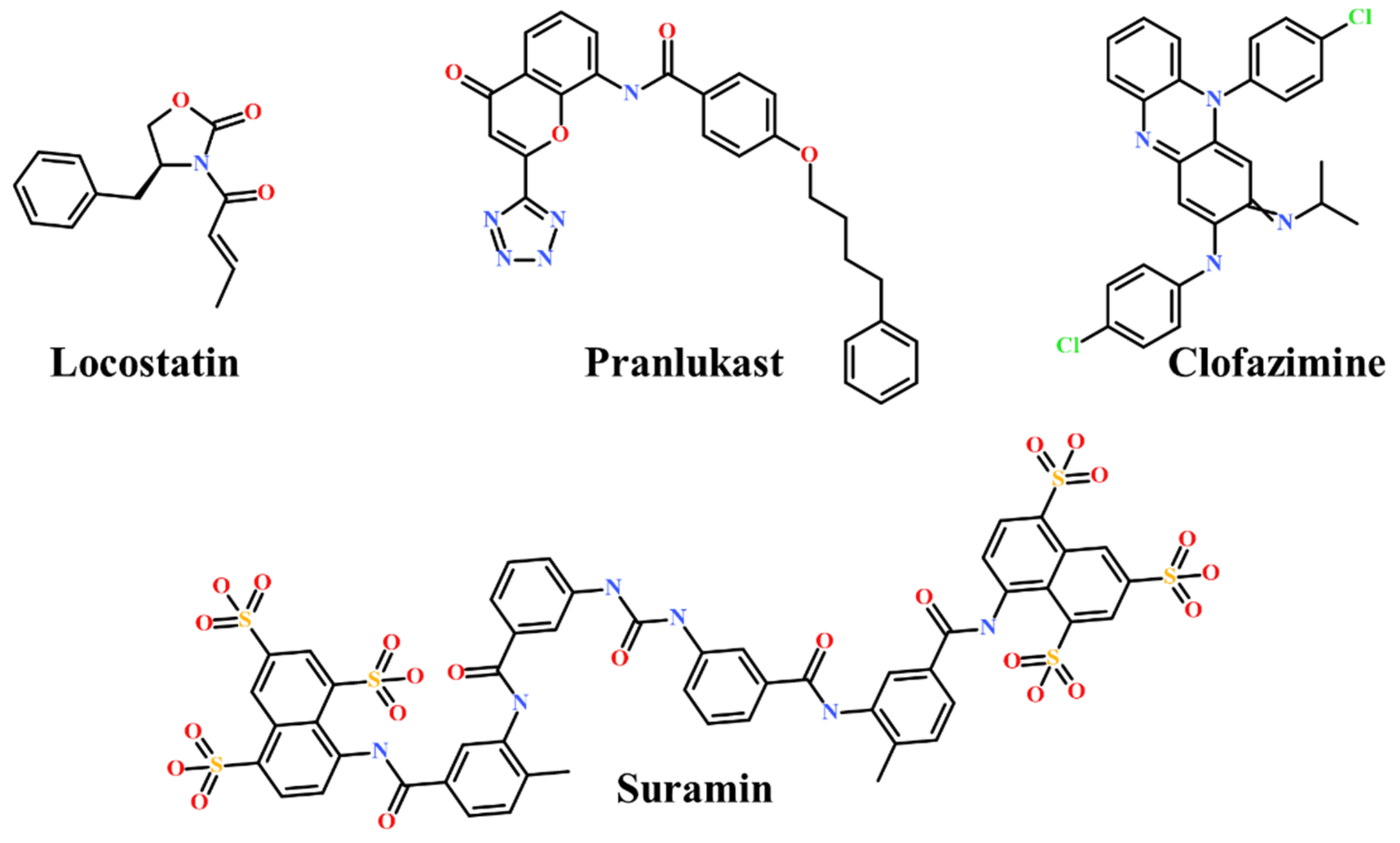
2.2. Drug-Like Marine-Derived Compounds from Virtual Screening
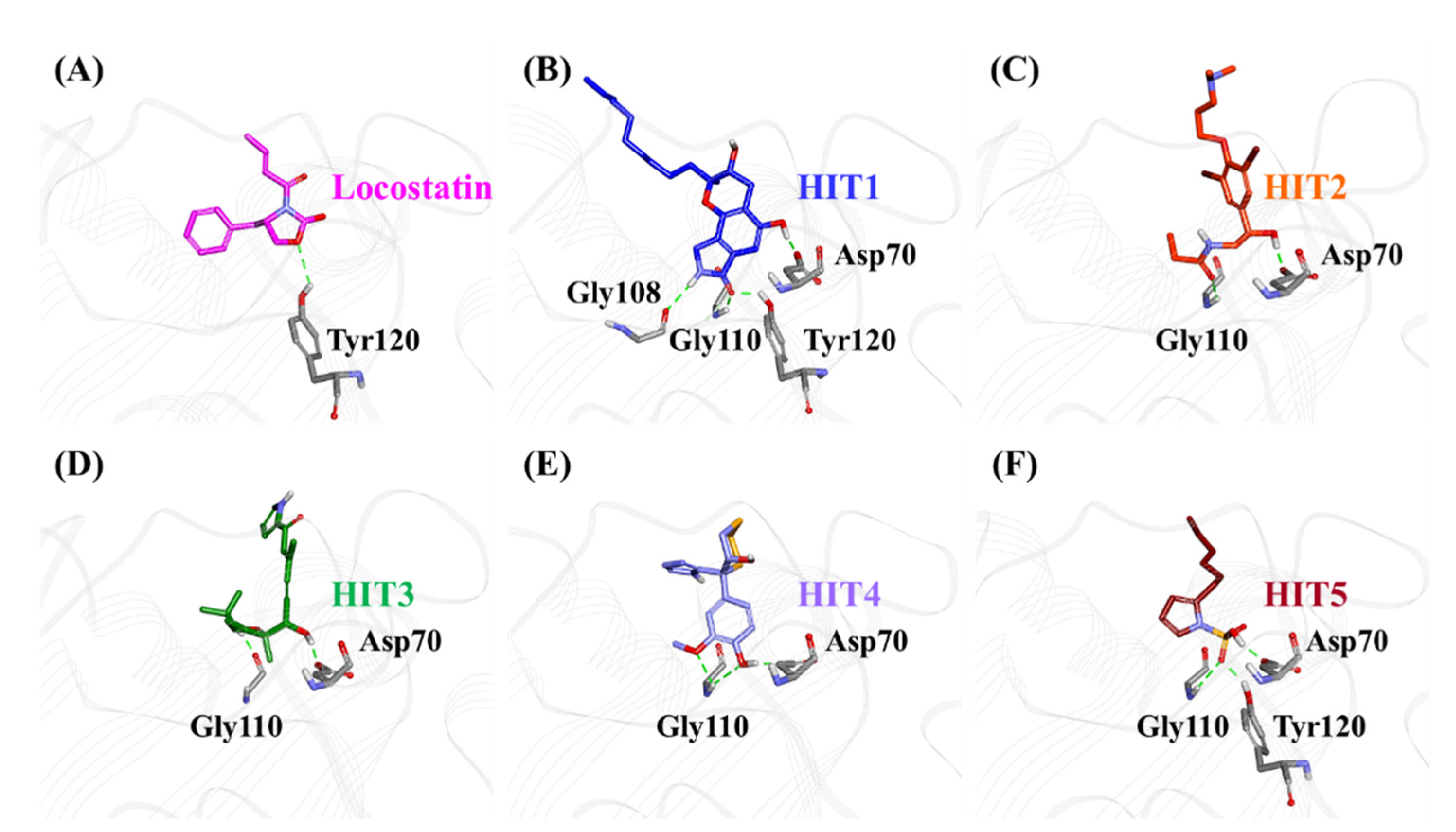
2.3. Molecular Docking of Screening-Derived Compounds with RKIP
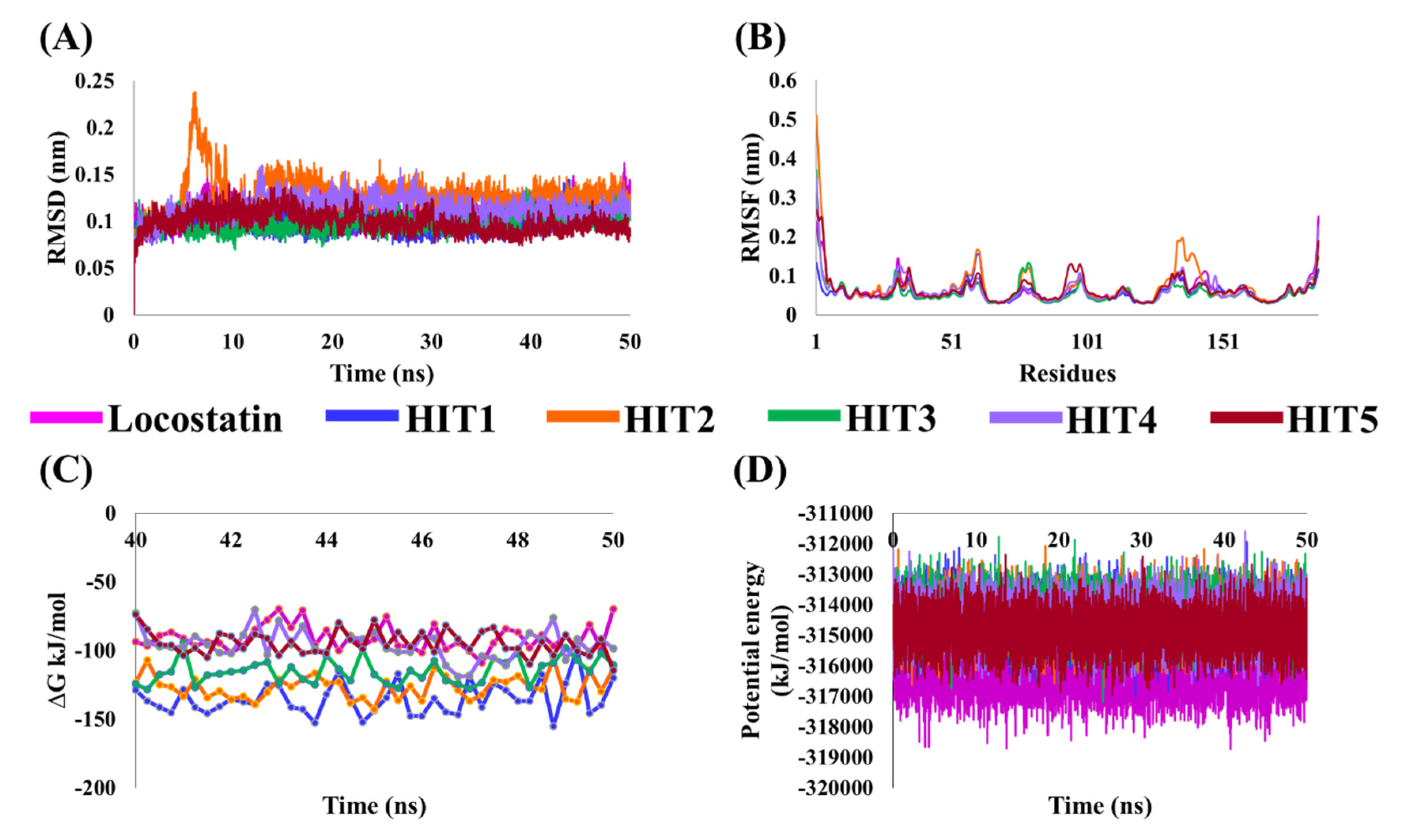
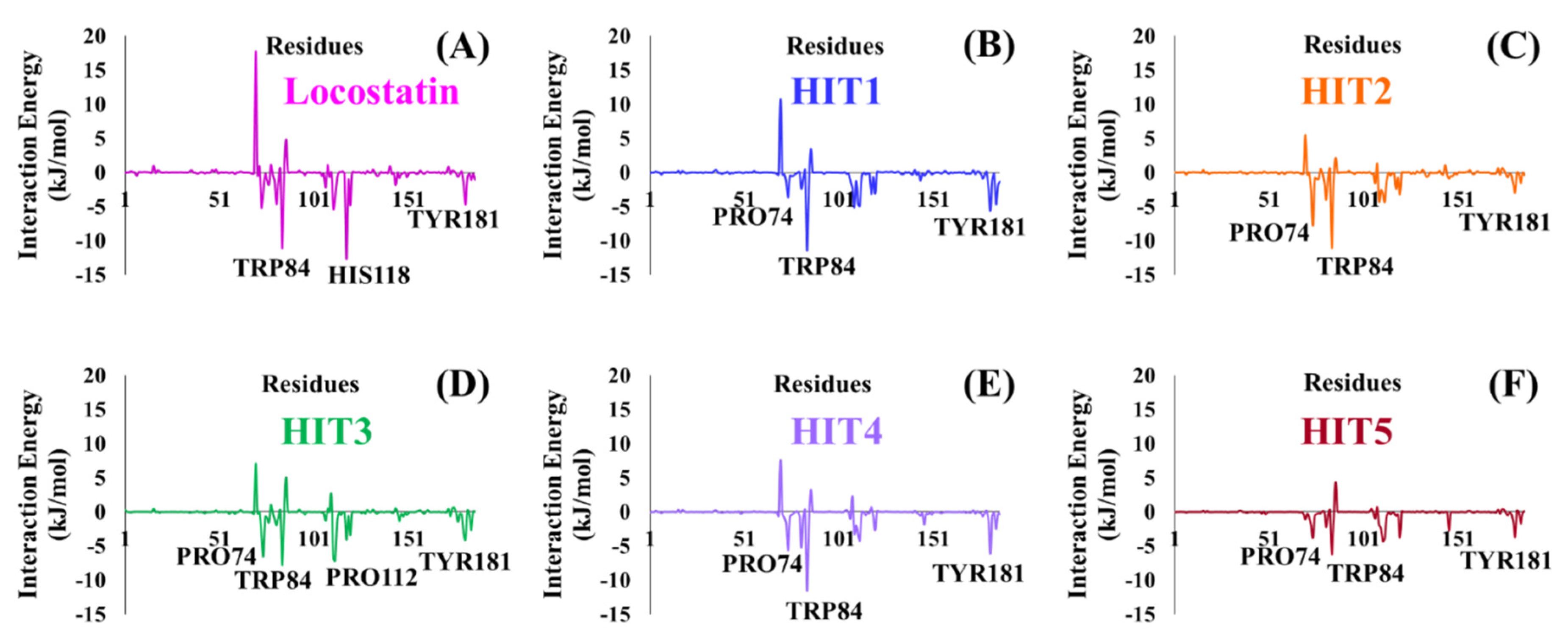
2.4. Molecular Dynamics Simulation Analyses
3. Discussion
4. Materials and Methods
4.1. Auto-Pharmacophore Model Generation
4.2. Virtual Screening of Marine-Derived Natural Products
4.3. Molecular Docking of Drug-Like Molecules with RKIP Ligand-Binding Pocket
4.4. Molecular Dynamics Simulation of Identified Marine-Derived Natural Products
4.5. Binding Free Energy Calculations of Identified Hits
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al-Mulla, F.; Bitar, M.S.; Taqi, Z.; Yeung, K.C. RKIP: Much more than Raf Kinase inhibitory protein. J. Cell. Physiol. 2013, 228, 1688–1702. [Google Scholar] [CrossRef]
- Keller, E.T.; Fu, Z.; Brennan, M. The role of Raf kinase inhibitor protein (RKIP) in health and disease. Biochem. Pharmacol. 2004, 68, 1049–1053. [Google Scholar]
- Parate, S.; Rampogu, S.; Lee, G.; Hong, J.C.; Lee, K.W. Exploring the Binding Interaction of Raf Kinase Inhibitory Protein With the N-Terminal of C-Raf Through Molecular Docking and Molecular Dynamics Simulation. Front. Mol. Biosci. 2021, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Gabriela-Freitas, M.; Pinheiro, J.; Raquel-Cunha, A.; Cardoso-Carneiro, D.; Martinho, O. Rkip as an inflammatory and immune system modulator: Implications in cancer. Biomolecules 2019, 9, 769. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, K.; Lohse, M.J.; Quitterer, U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003, 426, 574–579. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Li, Y.; Sarkar, F.H. The biological complexity of RKIP signaling in human cancers. Exp. Mol. Med. 2015, 47, e185. [Google Scholar] [CrossRef] [Green Version]
- Yesilkanal, A.E.; Rosner, M.R. Targeting raf kinase inhibitory protein regulation and function. Cancers 2018, 10, 306. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Kitagawa, Y.; Shen, R.; Shah, R.; Mehra, R.; Rhodes, D.; Keller, P.J.; Mizokami, A.; Dunn, R.; Chinnaiyan, A.M.; et al. Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate 2006, 66, 248–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinho, O.; Granja, S.; Jaraquemada, T.; Caeiro, C.; Miranda-Gonçalves, V.; Honavar, M.; Costa, P.; Damasceno, M.; Rosner, M.R.; Lopes, J.M.; et al. Downregulation of RKIP is associated with poor outcome and malignant progression in gliomas. PLoS ONE 2012, 7, e30769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagan, S.; Al-Mulla, F.; Mallon, E.; Oien, K.; Ferrier, R.; Gusterson, B.; Curto García, J.J.; Kolch, W. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin. Cancer Res. 2005, 11, 7392–7397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuierer, M.M.; Bataille, F.; Hagan, S.; Kolch, W.; Bosserhoff, A.K. Reduction in Raf kinase inhibitor protein expression is associated with increased Ras-extracellular signal-regulated kinase signaling in melanoma cell lines. Cancer Res. 2004, 64, 5186–5192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, L.Y.; Feng, F.; Zhao, Y.; Huang, M.Y.; Shao, Q.; Chen, C.; Sheng, H.; Chen, D.L.; Zeng, Z.L.; et al. Effect of Raf kinase inhibitor protein expression on malignant biological behavior and progression of colorectal cancer. Oncol. Rep. 2015, 34, 2106–2114. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Duan, G.; Zhao, C.; Gao, Y.; Liu, X.; Wang, Z.; Li, W.; Wang, K.; Wang, W. Reduced RKIP expression levels are associated with frequent non-small cell lung cancer metastasis and STAT3 phosphorylation and activation. Oncol. Lett. 2017, 13, 3039–3045. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Kim, G.Y.; Lim, S.J.; Kim, Y.W. Raf-1 kinase inhibitory protein expression in thyroid carcinomas. Endocr. Pathol. 2010, 21, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Yi, H.M.; Yi, H.; Qu, J.Q.; Zhu, J.F.; Li, L.N.; Xiao, T.; Zheng, Z.; Lu, S.S.; Xiao, Z.Q. Reduced RKIP enhances nasopharyngeal carcinoma radioresistance by increasing ERK and AKT activity. Oncotarget 2016, 7, 11463–11477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.J.; Holsinger, R.M.D.; McLean, C.A.; Tan, S.S.; Scott, H.S.; Cardamone, T.; Cappai, R.; Masters, C.L.; Li, Q.X. Decreased phosphatidylethanolamine binding protein expression correlates with Aβ accumulation in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol. Aging 2006, 27, 614–623. [Google Scholar] [CrossRef]
- Granovsky, A.E.; Rosner, M.R. Raf kinase inhibitory protein: A signal transduction modulator and metastasis suppressor. Cell Res. 2008, 18, 452–457. [Google Scholar] [CrossRef] [Green Version]
- Beshir, A.B.; Argueta, C.E.; Menikarachchi, L.C.; Gascón, J.A.; Fenteany, G. Locostatin disrupts association of Raf kinase inhibitor protein with binding proteins by modifying a conserved histidine residue in the ligand-binding pocket. For. Immunopathol. Dis. Therap. 2011, 2, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnitskaya, A.N.; Eddy, N.A.; Fenteany, G.; Gascón, J.A. Recognition and reactivity in the binding between Raf kinase inhibitor protein and its small-molecule inhibitor locostatin. J. Phys. Chem. B 2012, 116, 10176–10181. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wu, Z.; Luo, M.; Lin, D.; Guo, C. Pranlukast, a novel binding ligand of human Raf1 kinase inhibitory protein. Biotechnol. Lett. 2016, 38, 1375–1380. [Google Scholar] [CrossRef]
- Guo, C.; Chang, T.; Sun, T.; Wu, Z.; Dai, Y.; Yao, H.; Lin, D. Anti-leprosy drug Clofazimine binds to human Raf1 kinase inhibitory protein and enhances ERK phosphorylation. Acta Biochim. Biophys. Sin. 2018, 50, 1062–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Wu, Z.; Lin, W.; Xu, H.; Chang, T.; Dai, Y.; Lin, D. Suramin Targets the Conserved Ligand-Binding Pocket of Human Raf1 Kinase Inhibitory Protein. Molecules 2021, 26, 1151. [Google Scholar] [CrossRef]
- Mc Henry, K.T.; Ankala, S.V.; Ghosh, A.K.; Fenteany, G. A Non-Antibacterial Oxazolidinone Derivative that Inhibits Epithelial Cell Sheet Migration. ChemBioChem 2002, 3, 1105–1111. [Google Scholar] [CrossRef]
- Zhu, S.; Mc Henry, K.T.; Lane, W.S.; Fenteany, G. A chemical inhibitor reveals the role of Raf kinase inhibitor protein in cell migration. Chem. Biol. 2005, 12, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Bauer, R.A.; Wurst, J.M.; Tan, D.S. Expanding the range of “druggable” targets with natural product-based libraries: An academic perspective. Curr. Opin. Chem. Biol. 2010, 14, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, C. Marine Natural Products in Medicinal Chemistry. ACS Med. Chem. Lett. 2018, 9, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Shinde, P.; Banerjee, P.; Mandhare, A. Marine natural products as source of new drugs: A patent review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 283–309. [Google Scholar] [CrossRef] [PubMed]
- Matulja, D.; Wittine, K.; Malatesti, N.; Laclef, S.; Turks, M.; Markovic, M.K.; Ambrožić, G.; Marković, D. Marine Natural Products with High Anticancer Activities. Curr. Med. Chem. 2020, 27, 1243–1307. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.Y.; Choi, H. Natural products from marine organisms with neuroprotective activity in the experimental models of Alzheimer’s disease, Parkinson’s disease and ischemic brain stroke: Their molecular targets and action mechanisms. Arch. Pharm. Res. 2015, 38, 139–170. [Google Scholar] [CrossRef]
- Shemon, A.N.; Eves, E.M.; Clark, M.C.; Heil, G.; Granovsky, A.; Zeng, L.; Imamoto, A.; Koide, S.; Rosner, M.R. Raf kinase inhibitory protein protects cells against locostatin-mediated inhibition of migration. PLoS ONE 2009, 4, e6028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Zaravinos, A.; Bonavida, B.; Chatzaki, E.; Baritaki, S. RKIP: A key regulator in tumor metastasis initiation and resistance to apoptosis: Therapeutic targeting and impact. Cancers 2018, 10, 287. [Google Scholar] [CrossRef] [Green Version]
- Simister, P.C.; Burton, N.M.; Brady, R.L. Phosphotyrosine recognition by the raf kinase inhibitor protein. For. Immunopathol. Dis. Therap. 2011, 2, 59–70. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Tavel, L.; Jaquillard, L.; Karsisiotis, A.I.; Saab, F.; Jouvensal, L.; Brans, A.; Delmas, A.F.; Schoentgen, F.; Cadene, M.; Damblon, C. Ligand binding study of human PEBP1/RKIP: Interaction with nucleotides and raf-1 peptides evidenced by NMR and mass spectrometry. PLoS ONE 2012, 7, e36187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; de Guzman, F.S.; Gloer, J.B.; Shearer, C.A. Stachybotrins A and B: Novel Bioactive Metabolites from a Brackish Water Isolate of the Fungus Stachybotrys sp. J. Org. Chem. 1992, 57, 6700–6703. [Google Scholar] [CrossRef]
- Tilvi, S.; Rodrigues, C.; Naik, C.G.; Parameswaran, P.S.; Wahidhulla, S. New bromotyrosine alkaloids from the marine sponge Psammaplysilla purpurea. Tetrahedron 2004, 60, 10207–10215. [Google Scholar] [CrossRef]
- Barsby, T.; Kicklighter, C.E.; Hay, M.E.; Sullards, M.C.; Kubanek, J. Defensive 2-alkylpyrrole sulfamates from the marine annelid Cirriformia tentaculata. J. Nat. Prod. 2003, 66, 1110–1112. [Google Scholar] [CrossRef]
- Blunt, J.W.; Munro, M.H.G. Dictionary of Marine Natural Products with CD-ROM, 1st ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 2007; Available online: https://www.routledge.com/Dictionary-of-Marine-Natural-Products-with-CD-ROM/Blunt-Munro/p/book/9780849382161 (accessed on 26 May 2021).
- Macherla, V.R.; Liu, J.; Bellows, C.; Teisan, S.; Nicholson, B.; Lam, K.S.; Potts, B.C.M. Glaciapyrroles A, B, and C, pyrrolosesquiterpenes from a Streptomyces sp. isolated from an Alaskan marine sediment. J. Nat. Prod. 2005, 68, 780–783. [Google Scholar] [CrossRef] [PubMed]
- CAS 58115-31-4 Aurantiamide—BOC Sciences. Available online: https://www.bocsci.com/aurantiamide-cas-58115-31-4-item-189189.html (accessed on 26 May 2021).
- Pettit, G.R.; Butler, M.S.; Williams, M.D.; Filiatrault, M.J.; Pettit, R.K. Isolation and structure of hemibastadinols 1-3 from the Papua New Guinea marine sponge Ianthella basta. J. Nat. Prod. 1996, 59, 927–934. [Google Scholar] [CrossRef]
- Dictionary of Alkaloids with CD-ROM—Google Books. Available online: https://books.google.co.kr/books?id=mynNBQAAQBAJ&pg=PA1033&lpg=PA1033&dq=142677-12-1&source=bl&ots=JLw2aSbhuf&sig=ACfU3U1C5GJjiqtHw9UXnh_gnToAhdYrZQ&hl=en&sa=X&ved=2ahUKEwj6wZ-a_ubwAhUdx4sBHcR2DpgQ6AEwAXoECAIQAw#v=onepage&q=142677-12-1&f=false (accessed on 26 May 2021).
- Capon, R.J.; Skene, C.; Lacey, E.; Gill, J.H.; Wicker, J.; Heiland, K.; Friedel, T. Lorneamides A and B: Two New Aromatic Amides from a Southern Australian Marine Actinomycete. J. Nat. Prod. 2000, 63, 1682–1683. [Google Scholar] [CrossRef] [PubMed]
- Parate, S.; Kumar, V.; Lee, G.; Rampogu, S.; Hong, J.C.; Lee, K.W. Marine-Derived Natural Products as ATP-Competitive mTOR Kinase Inhibitors for Cancer Therapeutics. Pharmaceuticals 2021, 14, 282. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Genet. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Computational Investigation Identified Potential Chemical Scaffolds for Heparanase as Anticancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 5311. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Parate, S.; Yoon, S.; Lee, G.; Lee, K.W. Computational Simulations Identified Marine-Derived Natural Bioactive Compounds as Replication Inhibitors of SARS-CoV-2. Front. Microbiol. 2021, 12, 583. [Google Scholar] [CrossRef]
- Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Identification of Flavonoids as Putative ROS-1 Kinase Inhibitors Using Pharmacophore Modeling for NSCLC Therapeutics. Molecules 2021, 26, 2114. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Lopes, P.E.M.; Mackerell, A.D. Recent developments and applications of the CHARMM force fields. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, R.; Parate, S.; Yoon, S.; Lee, G.; Kim, D.; Lee, K.W. Identification of ACK1 Inhibitors as Anticancer Agents by using Computer-Aided Drug Designing. J. Mol. Struct. 2021, 130200. [Google Scholar] [CrossRef]
- Yang, C.Y.; Sun, H.; Chen, J.; Nikolovska-Coleska, Z.; Wang, S. Importance of ligand reorganization free energy in protein-ligand binding-affinity prediction. J. Am. Chem. Soc. 2009, 131, 13709–13721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Luo, S.; Cong, Y.; Zhong, S.; Zhang, J.Z.H.; Duan, L. An accurate free energy estimator: Based on MM/PBSA combined with interaction entropy for protein-ligand binding affinity. Nanoscale 2020, 12, 10737–10750. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacophore Model | Number of Features | Feature Set * |
|---|---|---|
| Pharmacophore hypothesis | 4 | 2HBA, RA, HyP |
| Compound No. | MNP ID (CAS No *) | Goldscore | Chemscore |
|---|---|---|---|
| 1 | 62541-09-7 | 67.72 | −33.37 |
| 2 | 799246-91-6 | 64.48 | −31.02 |
| 3 | 383191-01-3 | 60.85 | −28.18 |
| 4 | 313951-44-9 | 59.03 | −35.95 |
| 5 | 61897-90-3 | 58.61 | −27.03 |
| 6 | 302924-16-9 | 58.39 | −29.14 |
| 7 | 182806-09-3 | 58.37 | −27.80 |
| 8 | 587875-53-4 | 57.76 | −28.71 |
| 9 | 142677-12-1 | 57.18 | −35.13 |
| 10 | 144385-02-4 | 57.07 | −30.29 |
| 11 | 853885-48-0 | 56.93 | −27.96 |
| 12 | 853885-46-8 | 56.04 | −34.01 |
| 13 | 58115-31-4 | 55.01 | −37.15 |
| 14 | 133812-16-5 (REF) | 48.64 | −26.48 |
| Hit No. | MNP ID (CAS No *) | Van Der Waals (kJ/mol) | Electrostatic (kJ/mol) | Polar Solvation (kJ/mol) | SASA Energy (kJ/mol) | BFE Scores ∆Gbind (kJ/mol) |
|---|---|---|---|---|---|---|
| HIT1 | 144385-02-4 | −171.724 ± 13.242 | −89.779 ± 8.171 | 143.371 ± 8.006 | −17.151 ± 1.008 | −135.283 ± 11.815 |
| HIT2 | 799246-91-6 | −159.666 ± 9.645 | −66.487 ± 8.367 | 115.080 ± 11.961 | −15.523 ± 0.756 | −126.597 ± 8.883 |
| HIT3 | 853885-46-8 | −152.922 ± 10.576 | −64.325 ± 7.461 | 117.843 ± 8.005 | −15.684 ± 0.925 | −115.088 ± 9.005 |
| HIT4 | 383191-01-3 | −144.389 ± 13.393 | −68.213 ± 8.692 | 131.246 ± 6.782 | −14.093 ± 0.641 | −95.450 ± 10.777 |
| HIT5 | 587875-53-4 | −118.918 ± 10.507 | −78.010 ± 7.488 | 114.660 ± 4.794 | −12.315 ± 0.793 | −94.582 ± 8.703 |
| HIT6 | 133812-16-5 (REF) | −149.624 ± 7.721 | −62.839 ± 5.691 | 134.966 ± 6.467 | −13.413 ± 0.628 | −90.909 ± 9.155 |
| HIT/CAS No * | Chemical Name (Source) | Reference | Molecular Structure |
|---|---|---|---|
| HIT1 (144385-02-4) | Stachybotrin B (Stachybotrys sp.) | [39] |  |
| HIT2 (799246-91-6) | Purpurealidin G (Psammaplysilla purpurea) | [40] |  |
| HIT3 (853885-46-8) | Glaciapyrrole A (Streptomyces sp.) | [42,43] |  |
| HIT4 (383191-01-3) | (Hypsistozoa fasmeriana) | [42] |  |
| HIT5 (587875-53-4) | 2-Hexylpyrrole sulfamate (Cirriformia tentaculata) | [41,42] |  |
| 58115-31-4 | Aurantiamide | [44] |  |
| 182806-09-3 | Hemibastadinol 1 (Ianthella basta) | [45] |  |
| 61897-90-3 | Fumitremorgin H. (Aspergillus fumigatus) | [42] |  |
| 142677-12-1 | (Chondria sp.) | [46] |  |
| 313951-44-9 | Lorneamide A | [46,47] |  |
| 302924-16-9 | Secobipinnatin J (Pseudopterogorgia bipinnata) | [42] |  |
| 853885-48-0 | Glaciapyrrole B (Streptomyces sp.) | [42,43] |  |
| 62541-09-7 | Dehydrocoelenterazine (Watasenia dehydropreluciferin) | [42] |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Investigation of Marine-Derived Natural Products as Raf Kinase Inhibitory Protein (RKIP)-Binding Ligands. Mar. Drugs 2021, 19, 581. https://doi.org/10.3390/md19100581
Parate S, Kumar V, Hong JC, Lee KW. Investigation of Marine-Derived Natural Products as Raf Kinase Inhibitory Protein (RKIP)-Binding Ligands. Marine Drugs. 2021; 19(10):581. https://doi.org/10.3390/md19100581
Chicago/Turabian StyleParate, Shraddha, Vikas Kumar, Jong Chan Hong, and Keun Woo Lee. 2021. "Investigation of Marine-Derived Natural Products as Raf Kinase Inhibitory Protein (RKIP)-Binding Ligands" Marine Drugs 19, no. 10: 581. https://doi.org/10.3390/md19100581
APA StyleParate, S., Kumar, V., Hong, J. C., & Lee, K. W. (2021). Investigation of Marine-Derived Natural Products as Raf Kinase Inhibitory Protein (RKIP)-Binding Ligands. Marine Drugs, 19(10), 581. https://doi.org/10.3390/md19100581