
Recent Developments on the Synthesis and Bioactivity of Ilamycins/Rufomycins and Cyclomarins, Marine Cyclopeptides That Demonstrate Anti-Malaria and Anti-Tuberculosis Activity


Abstract
1. Introduction
2. Discovery of Anti-Tubercular Cycloheptapeptides
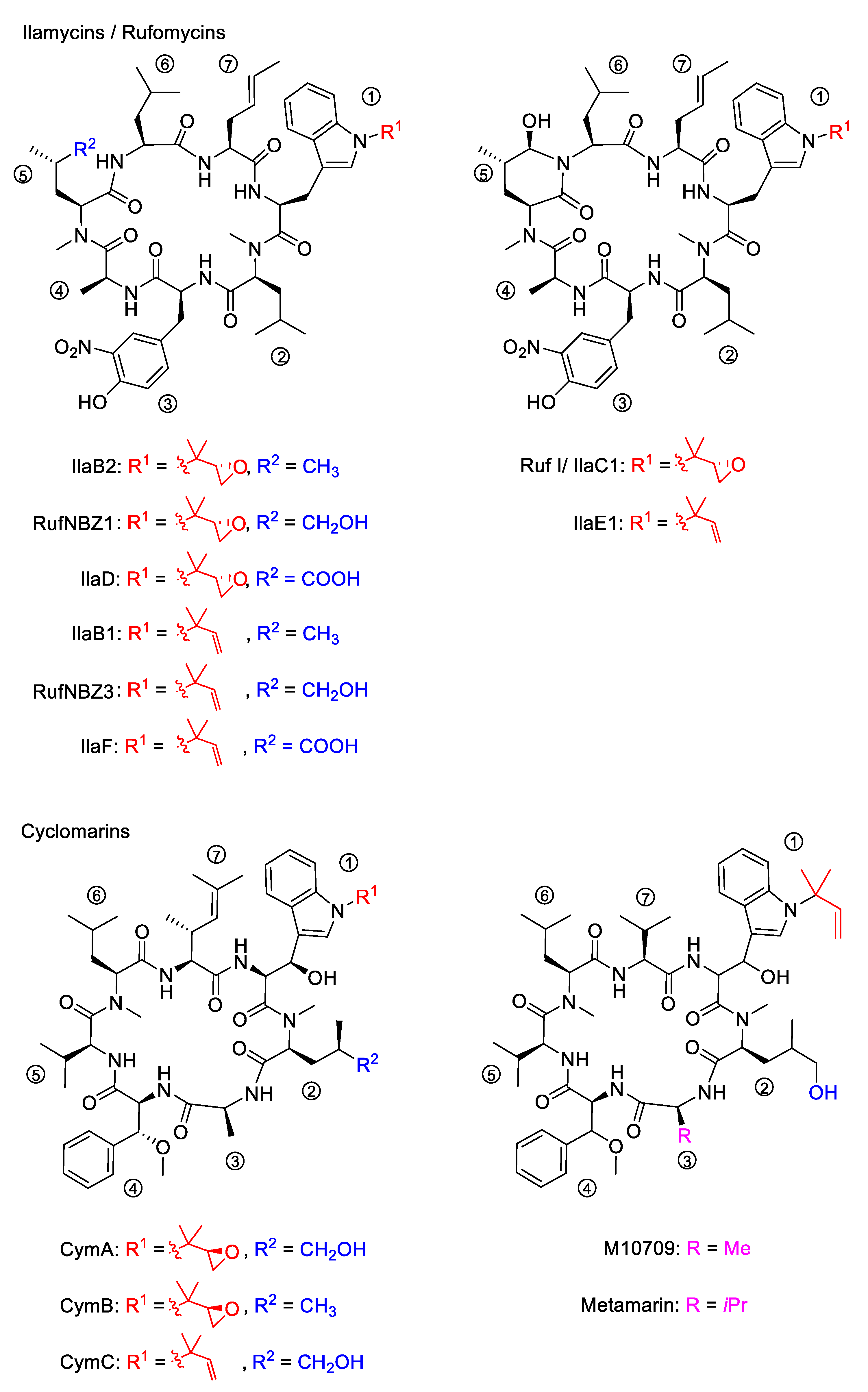
2.1. Discovery of the Ilamycins/Rufomycins
2.2. Discovery of the Cyclomarins
3. Biosynthesis of Anti-Tubercular Cycloheptapeptides
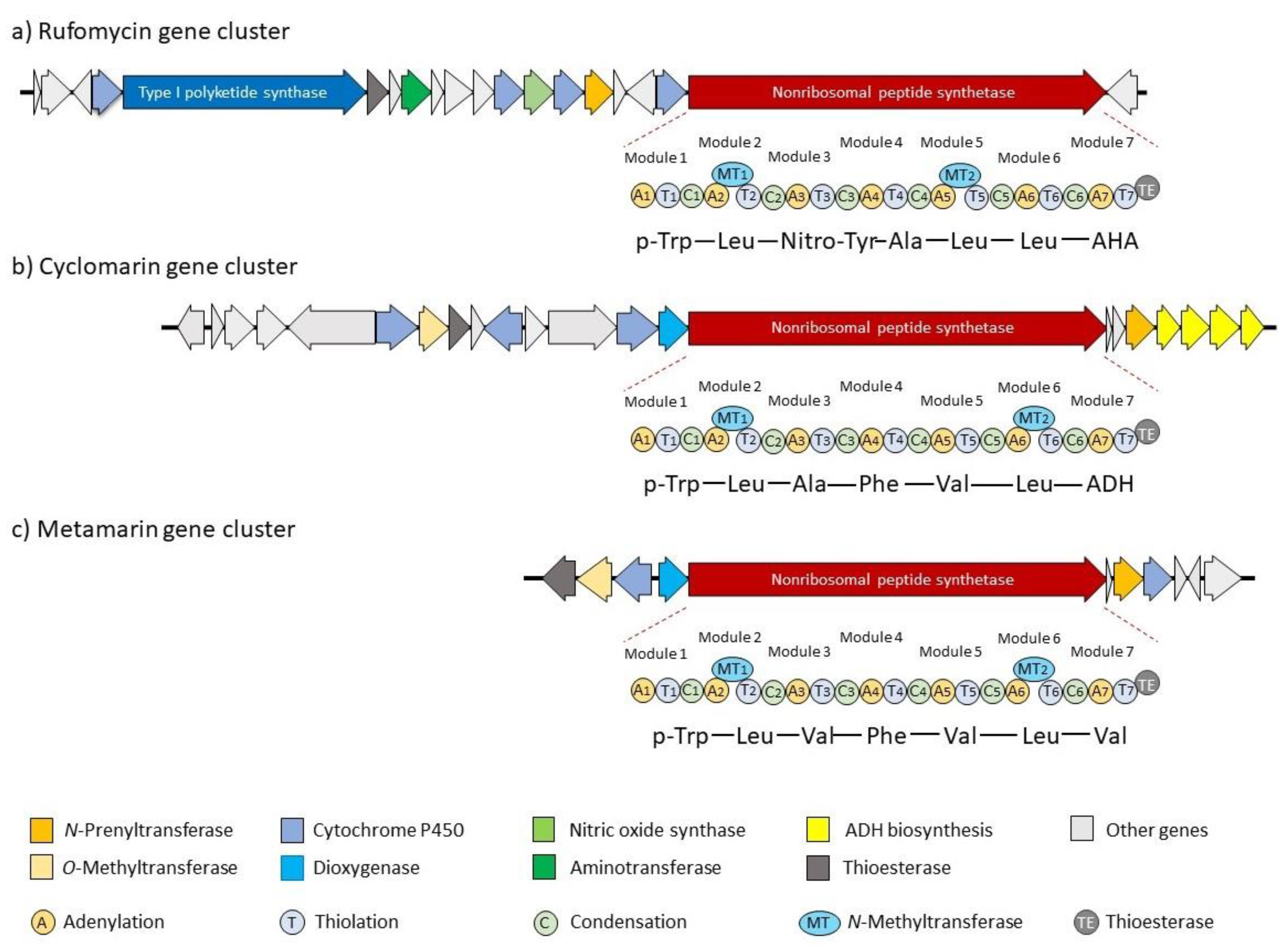
3.1. Biosynthesis of the Ilamycins/Rufomycins
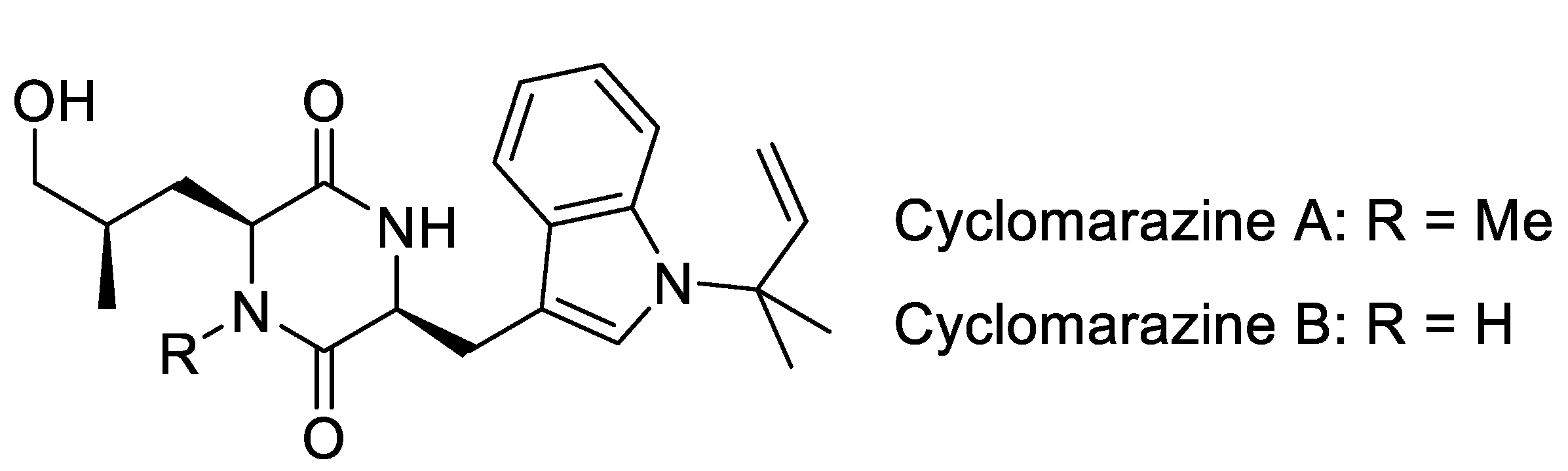
3.2. Biosynthesis of Cyclomarins
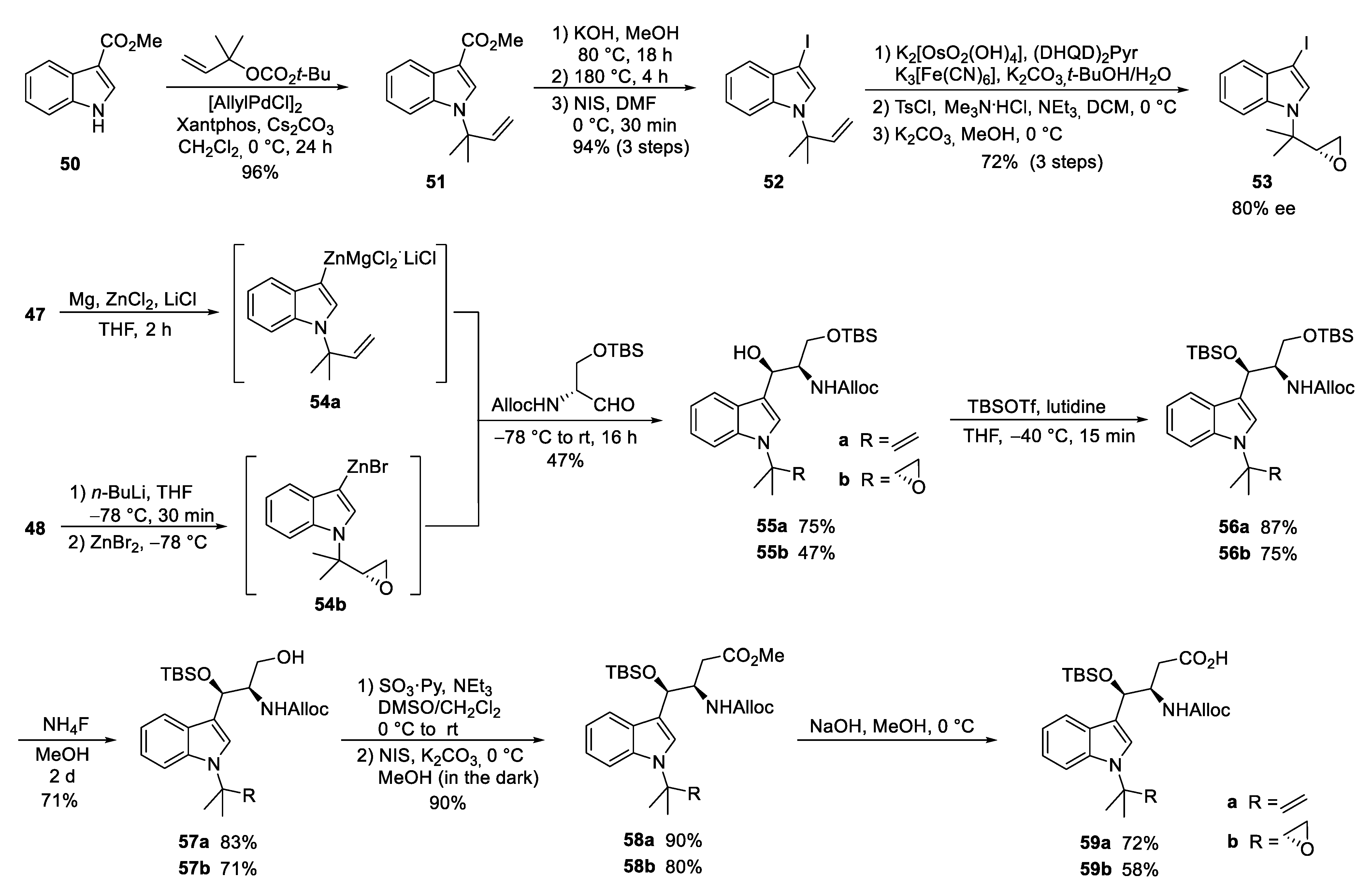
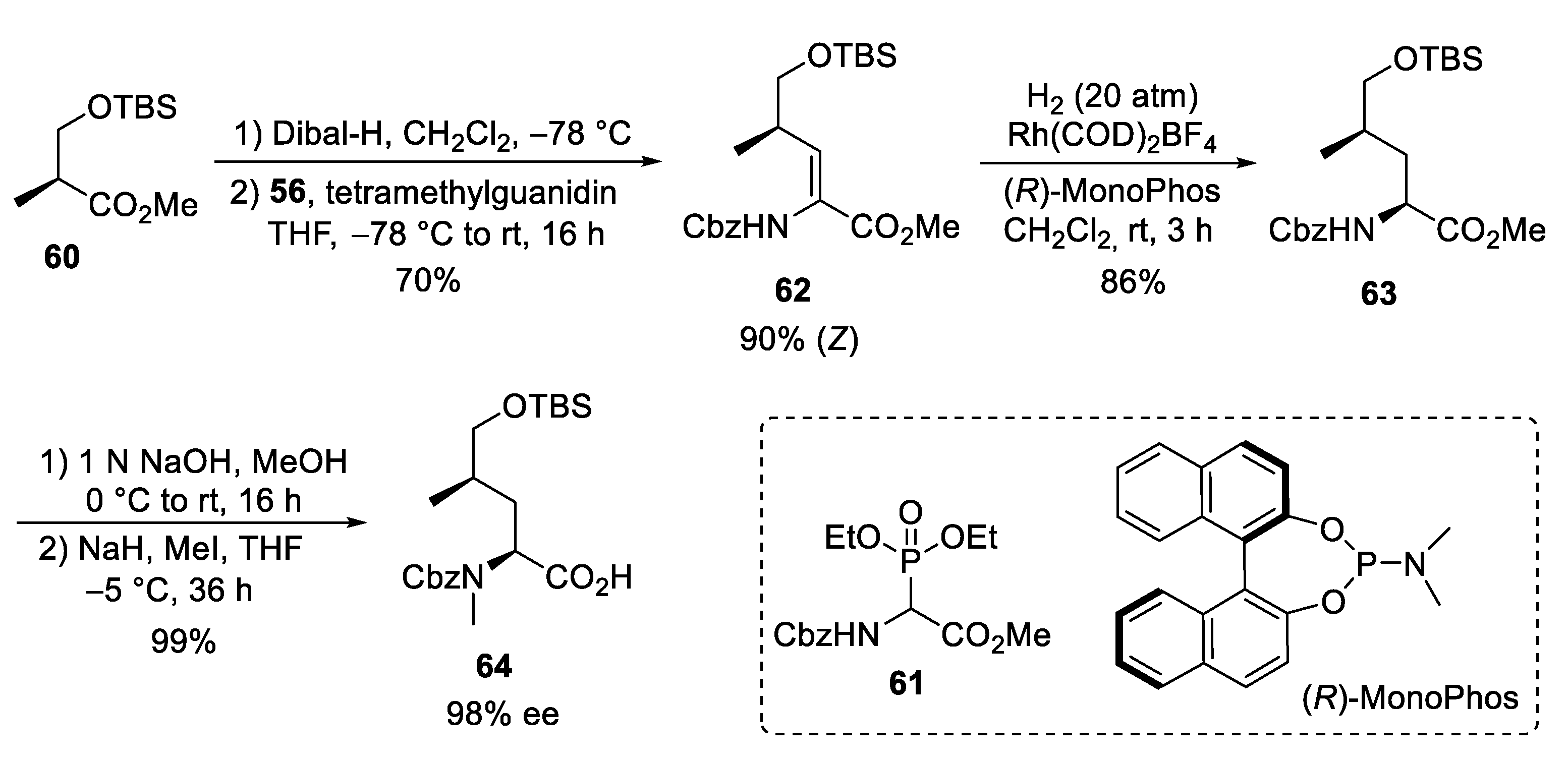
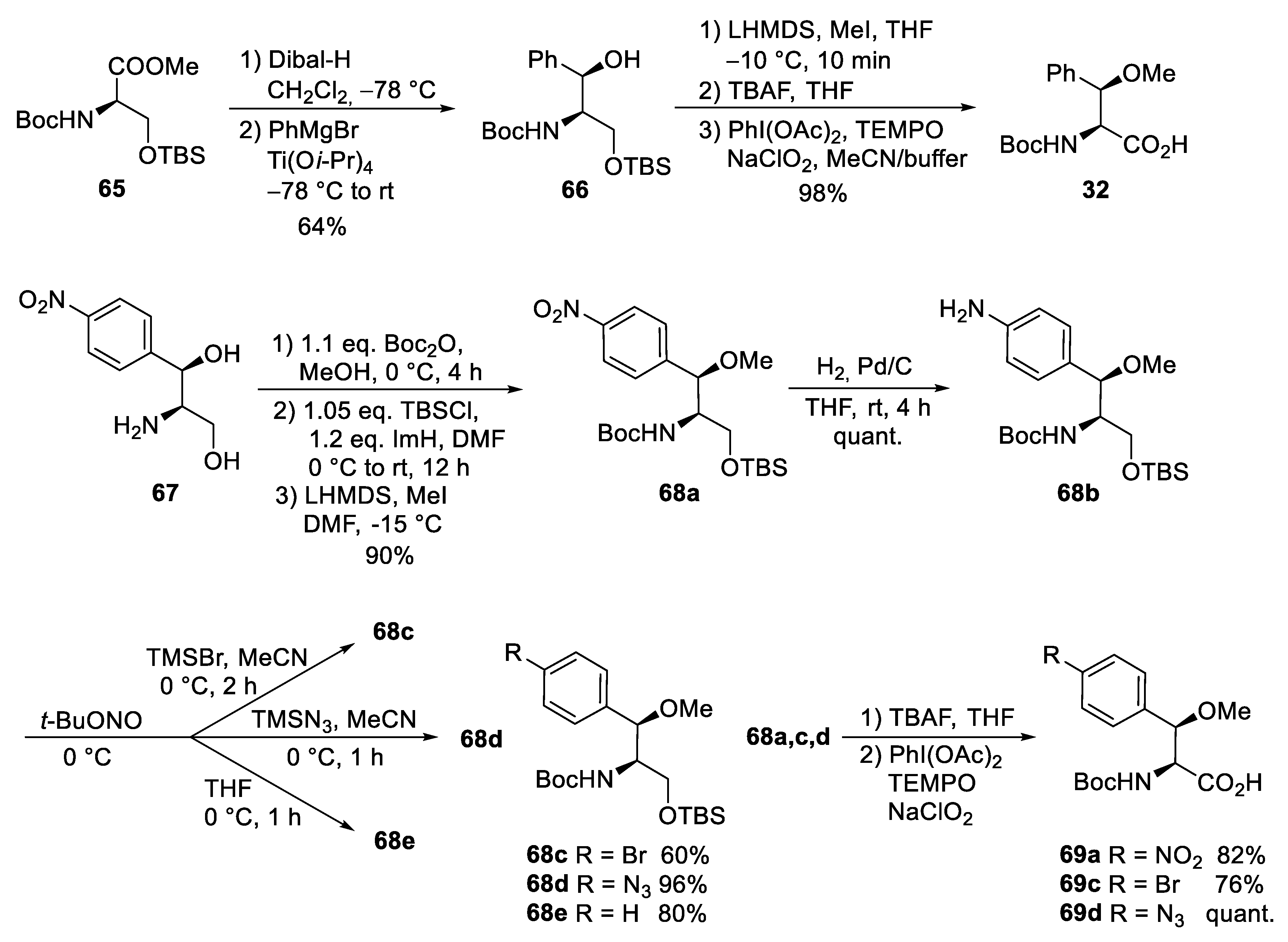
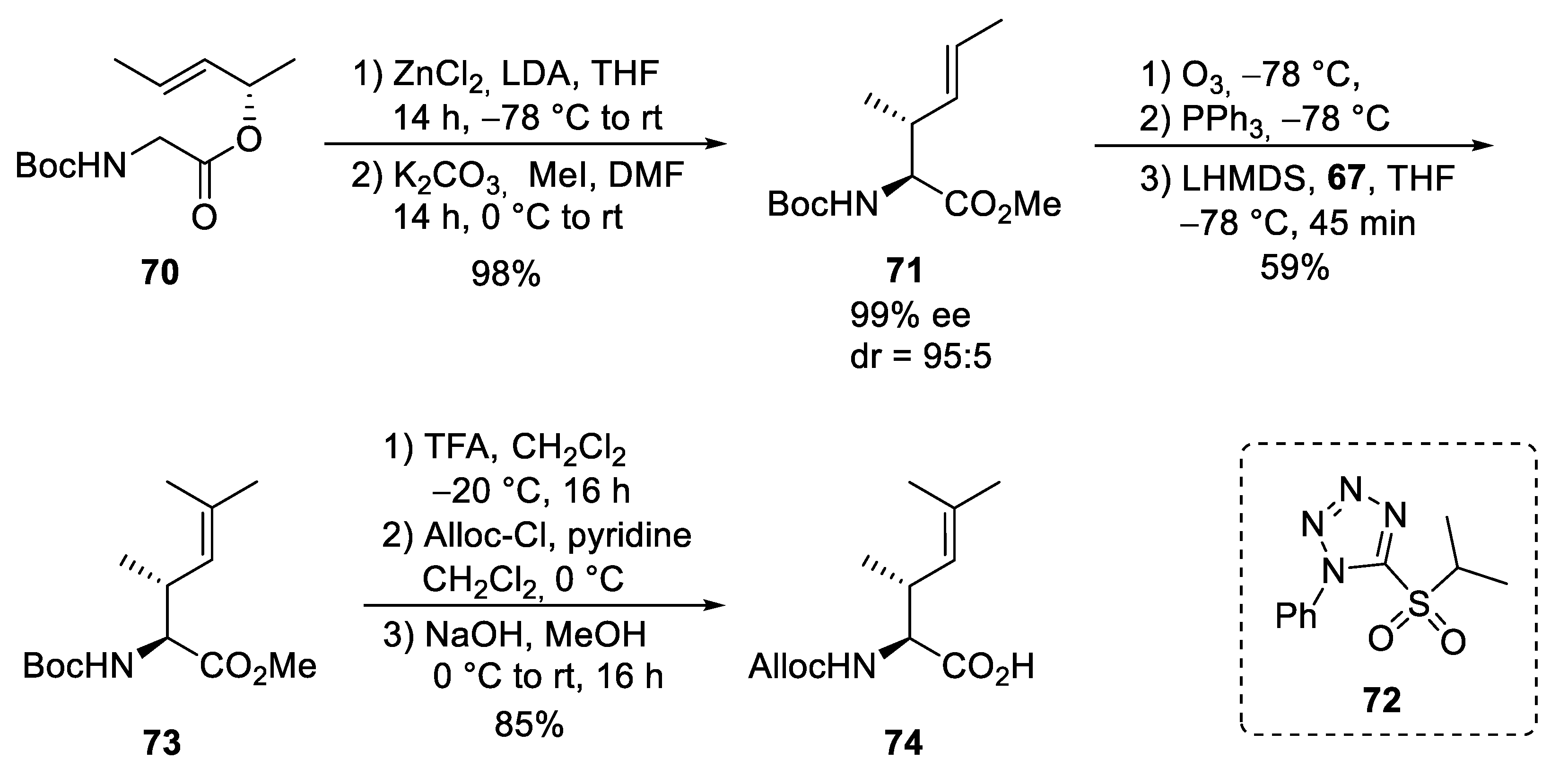
4. Total Syntheses of Marine Cycloheptapeptides
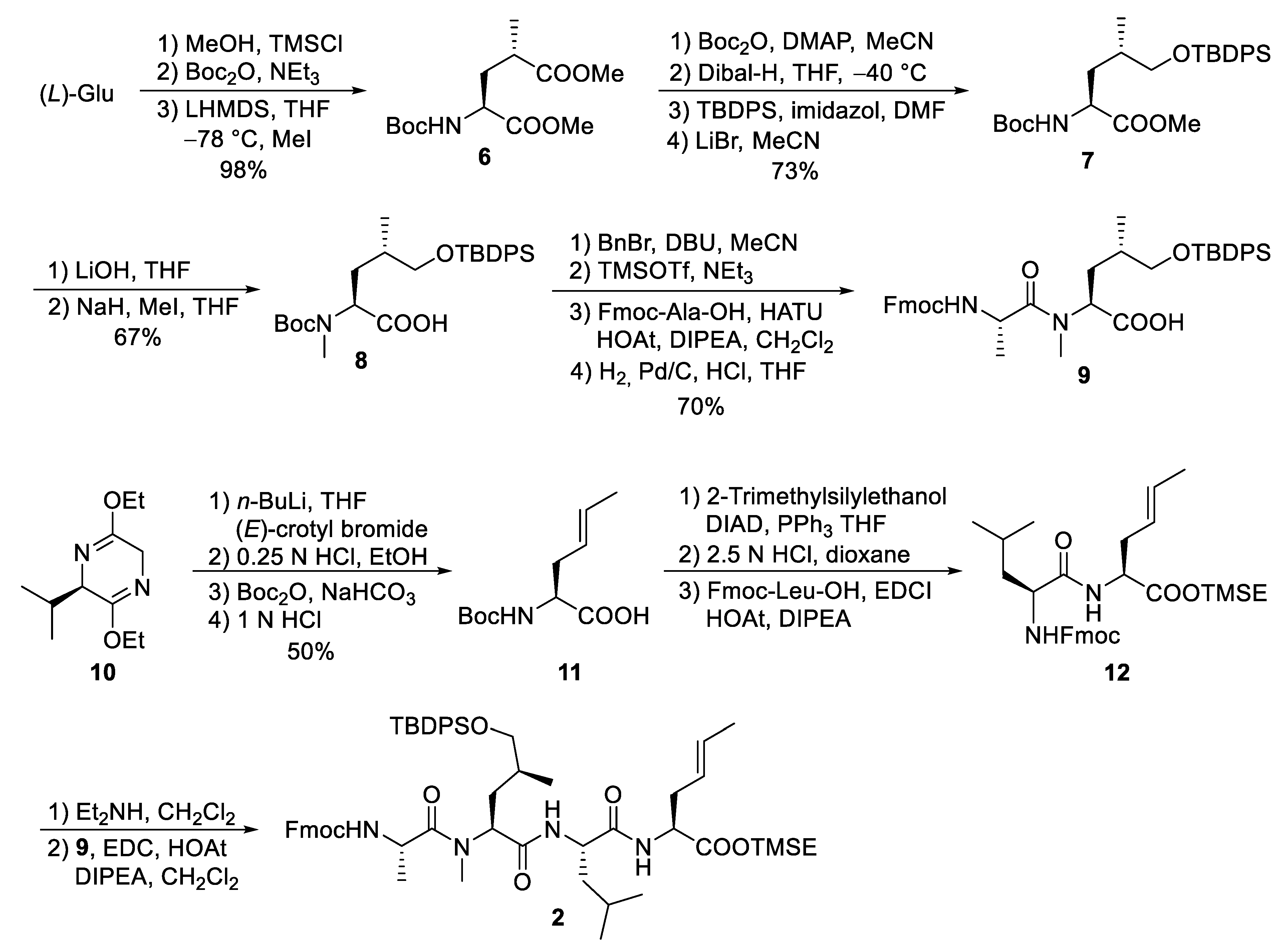
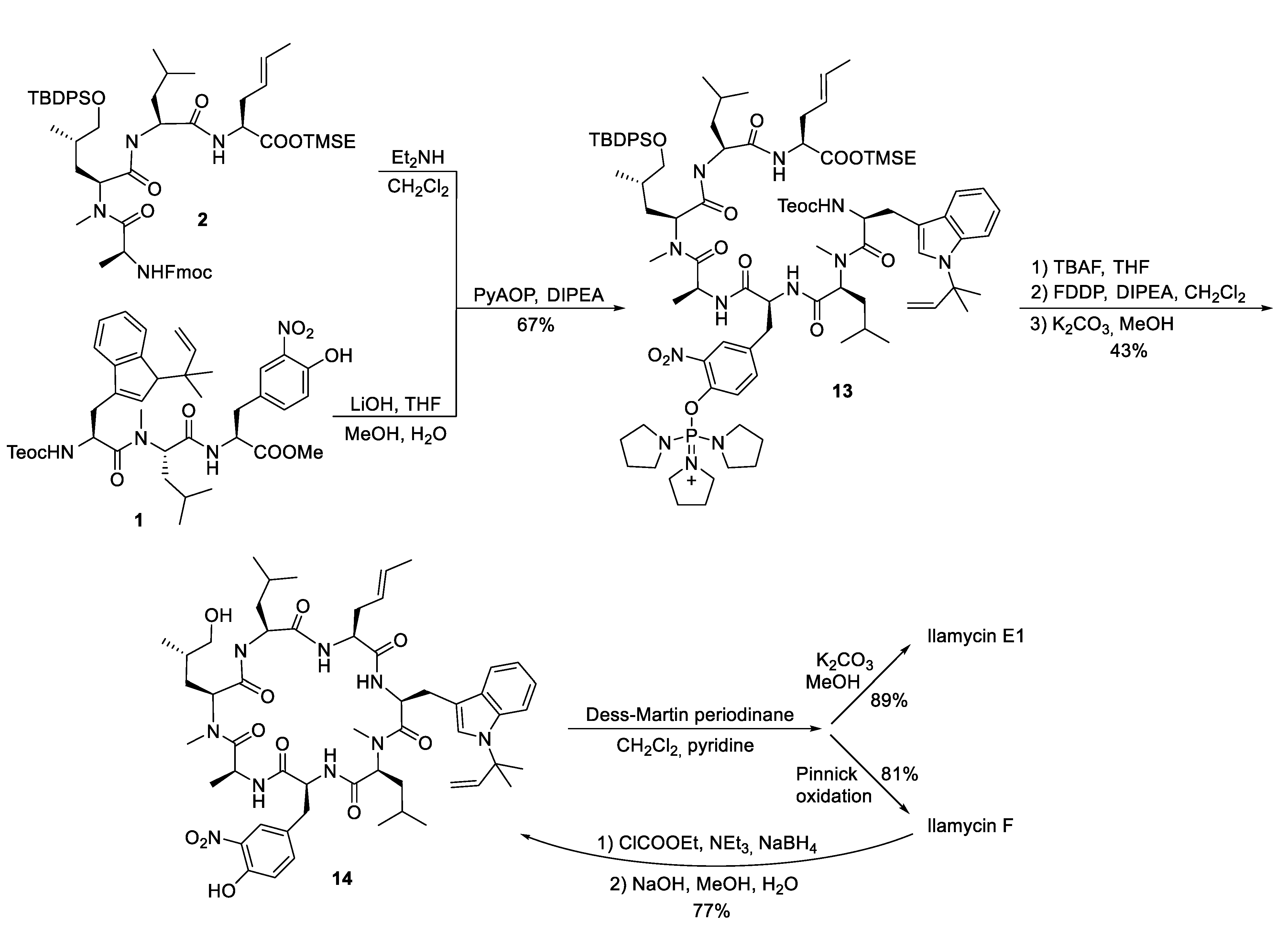
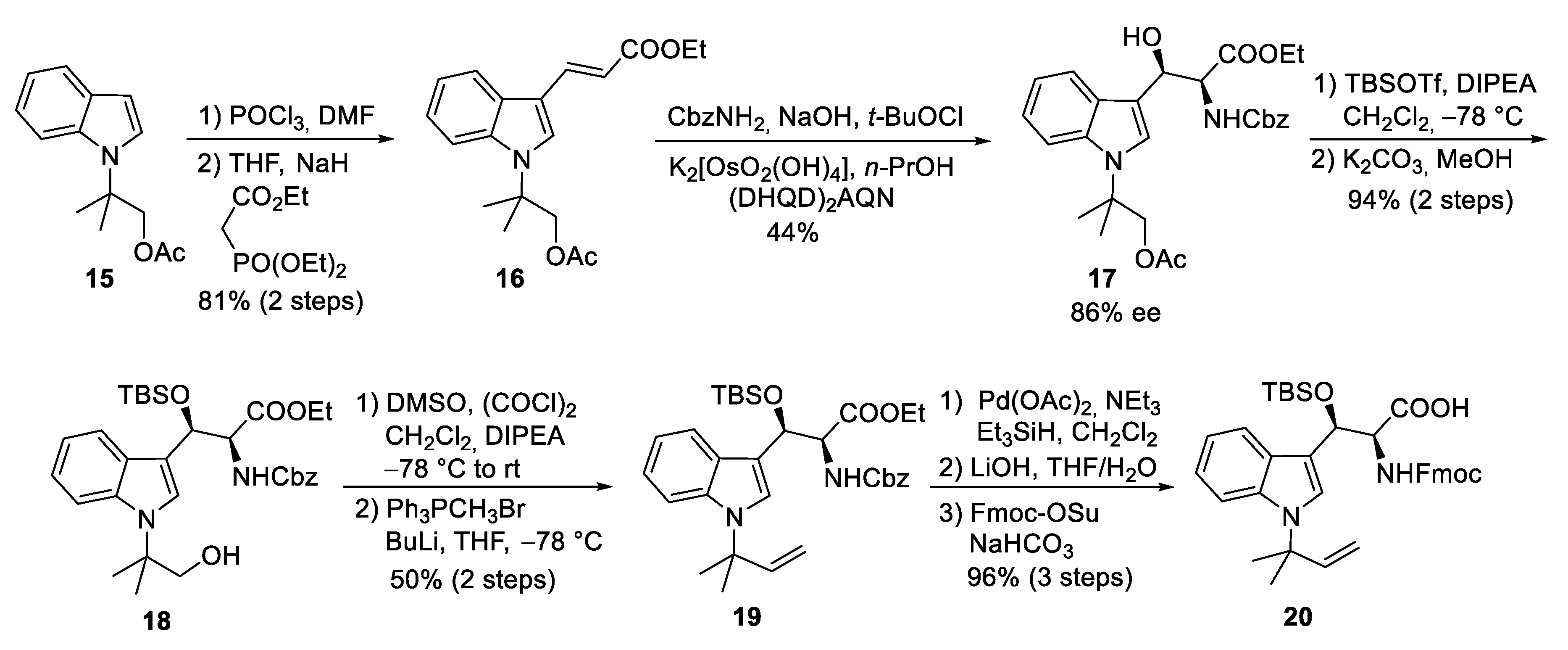
4.1. Total Synthesis of Ilamycins/Rufomycins
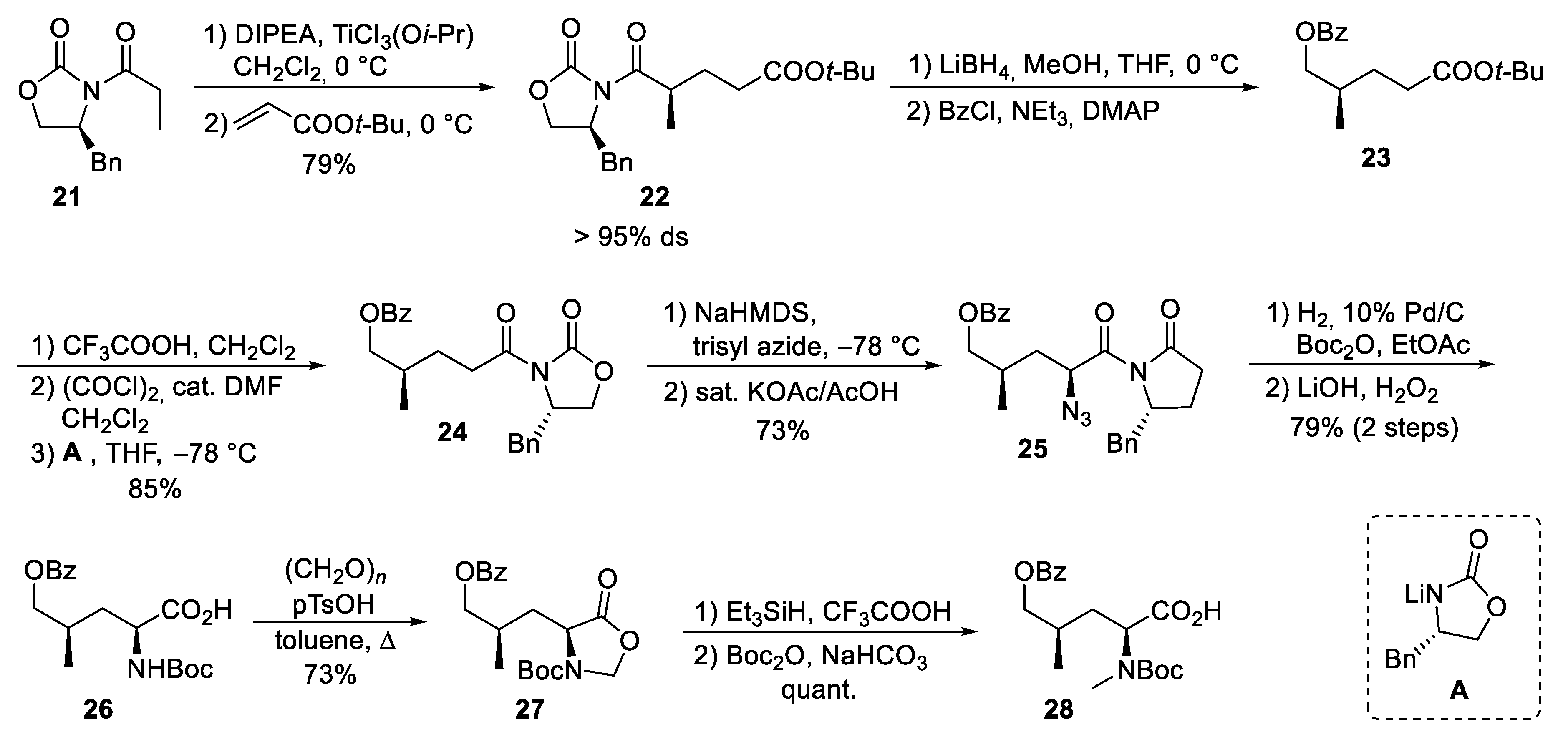
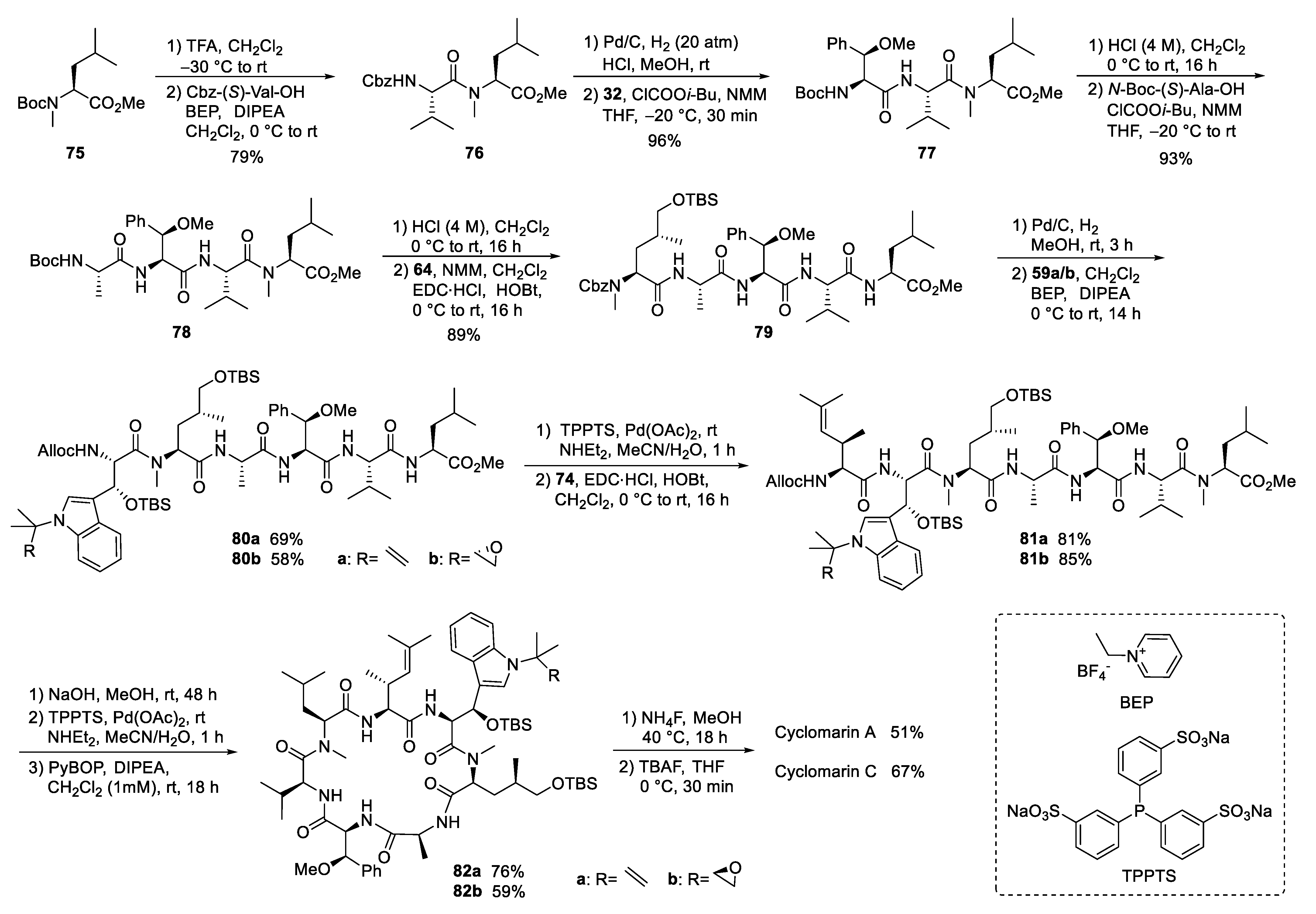
4.2. Total Synthesis of Cyclomarins
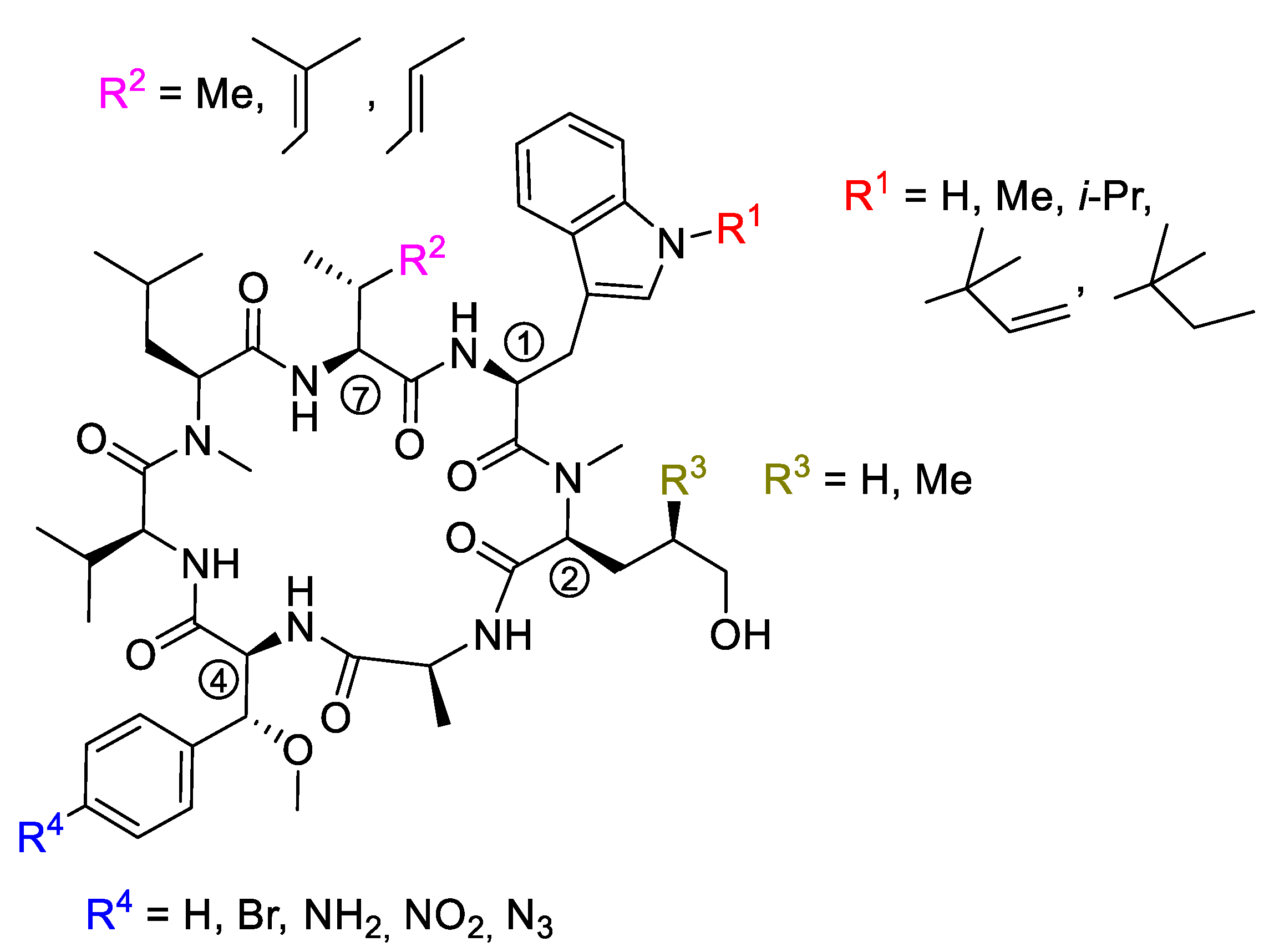
5. Syntheses of Cyclomarin Derivatives
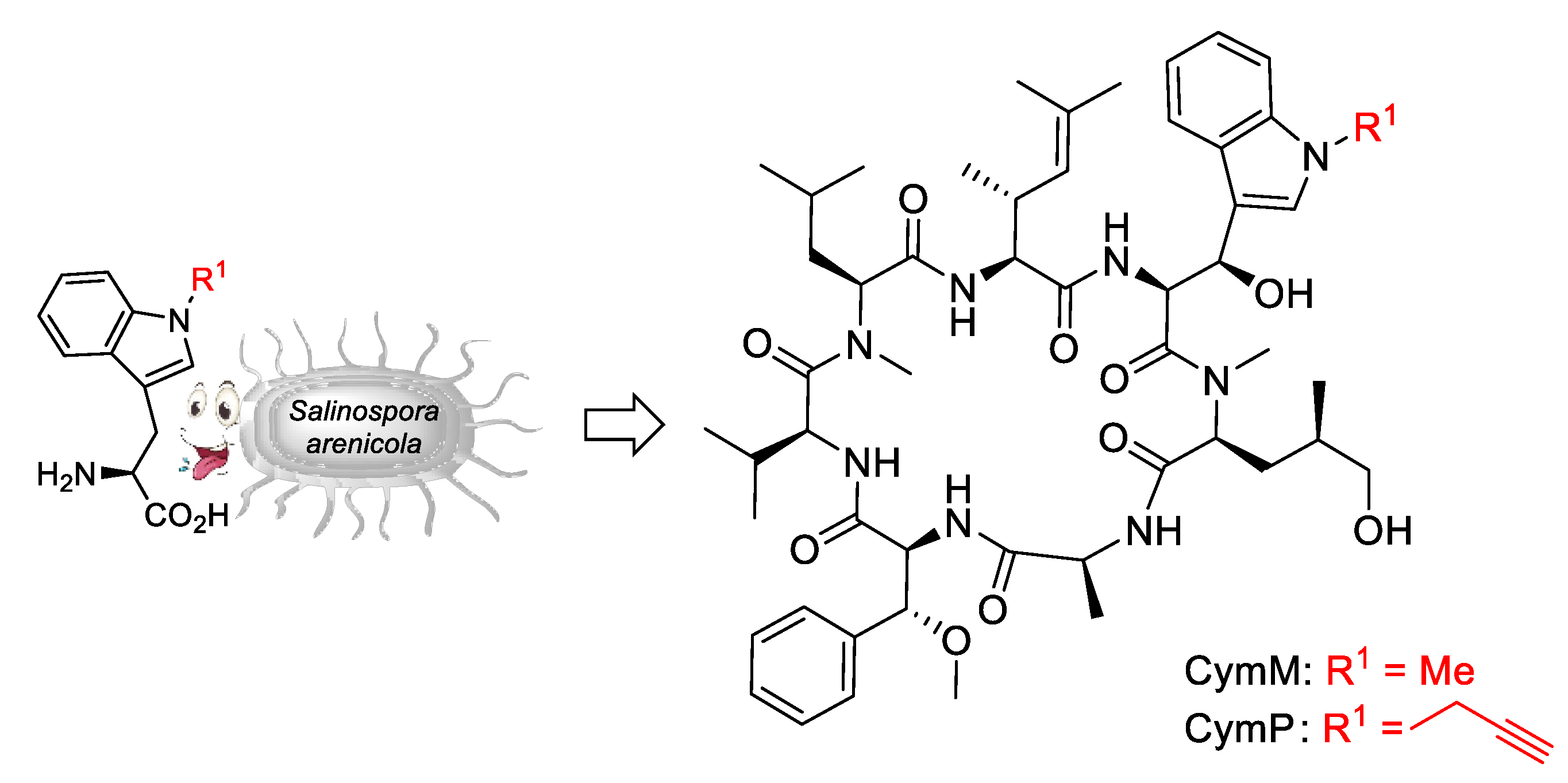
5.1. Mutasyntheses of Cyclomarin Derivatives
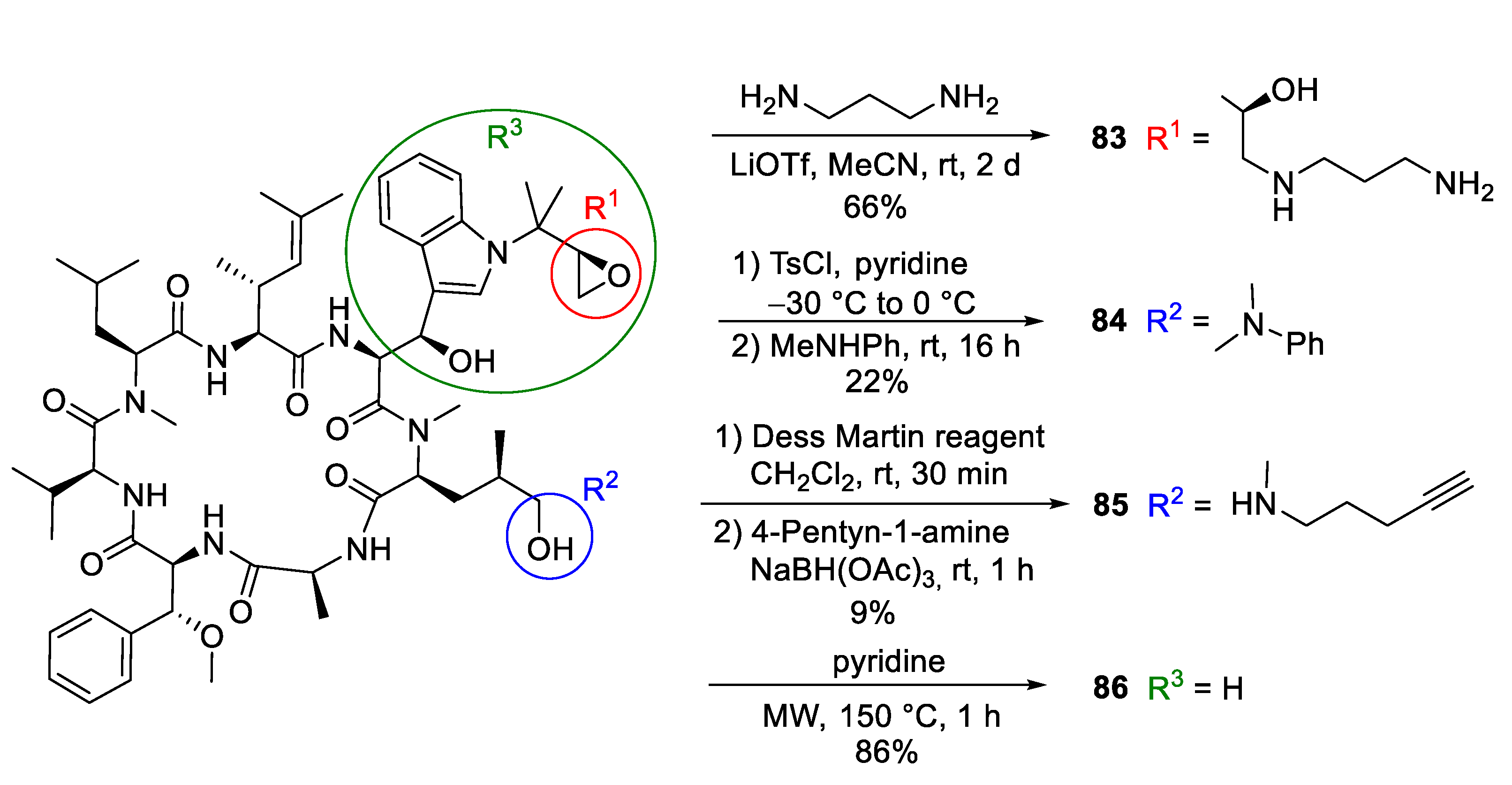
5.2. Semisyntheses of Cyclomarin Derivatives
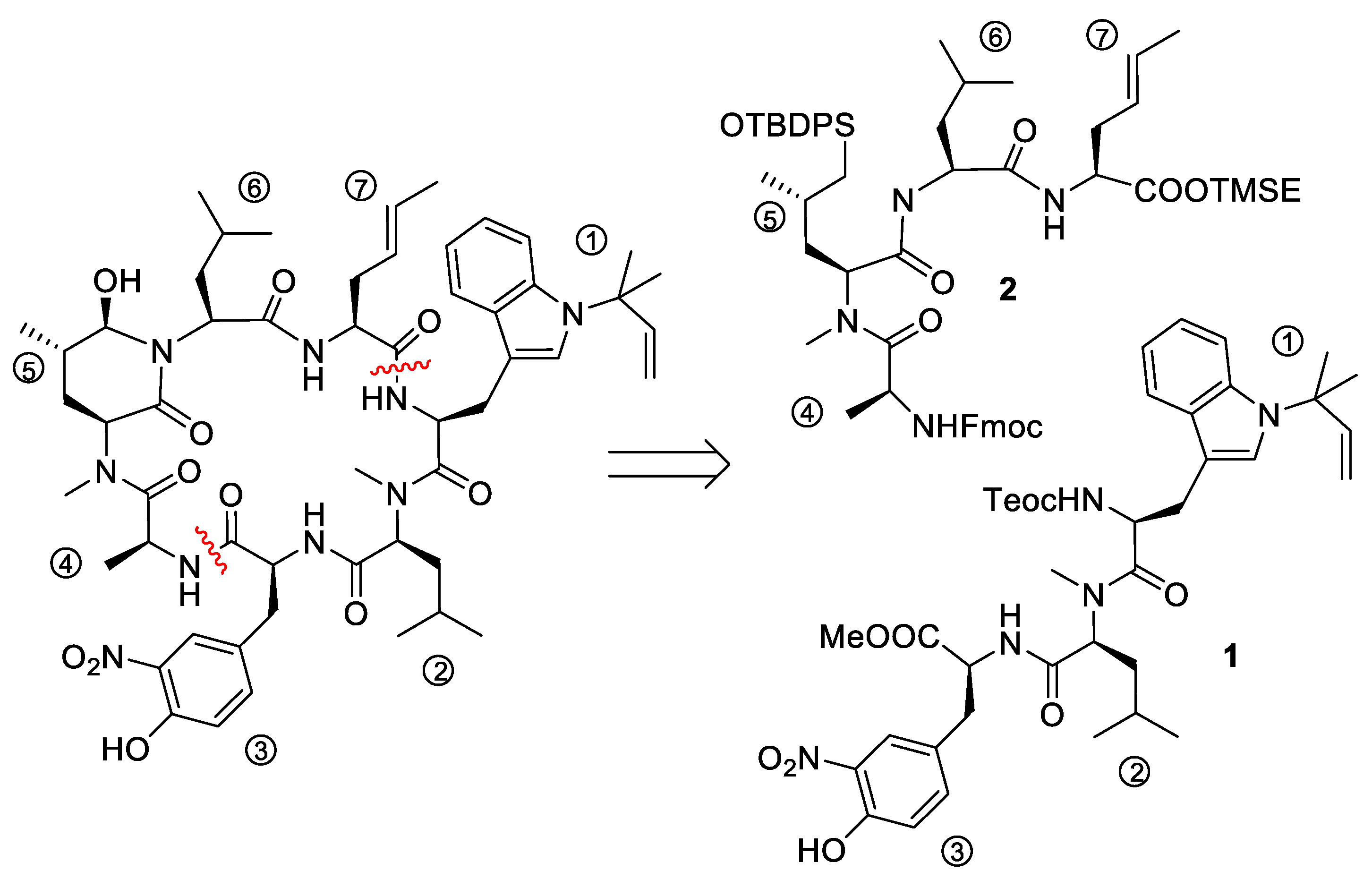
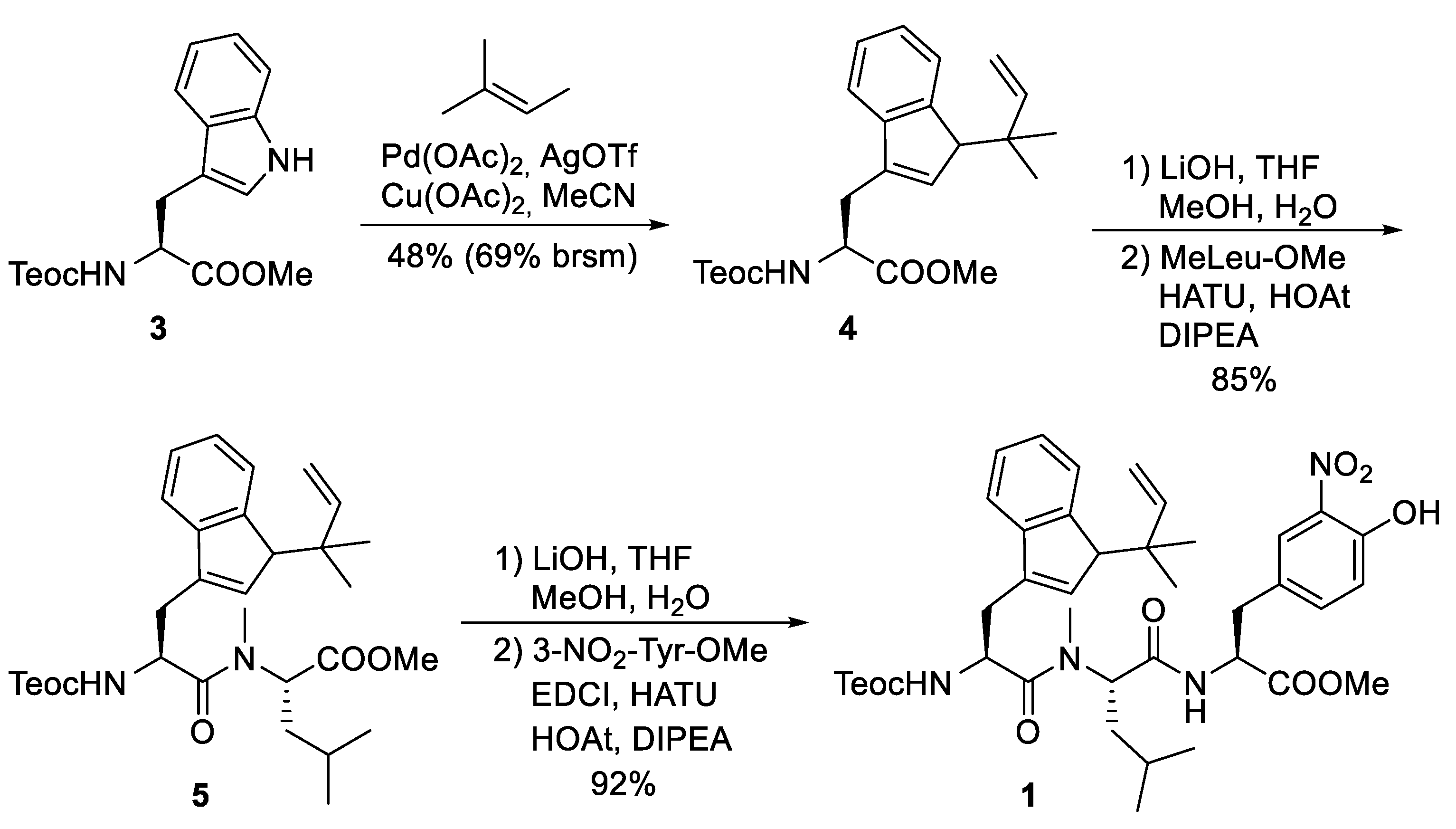
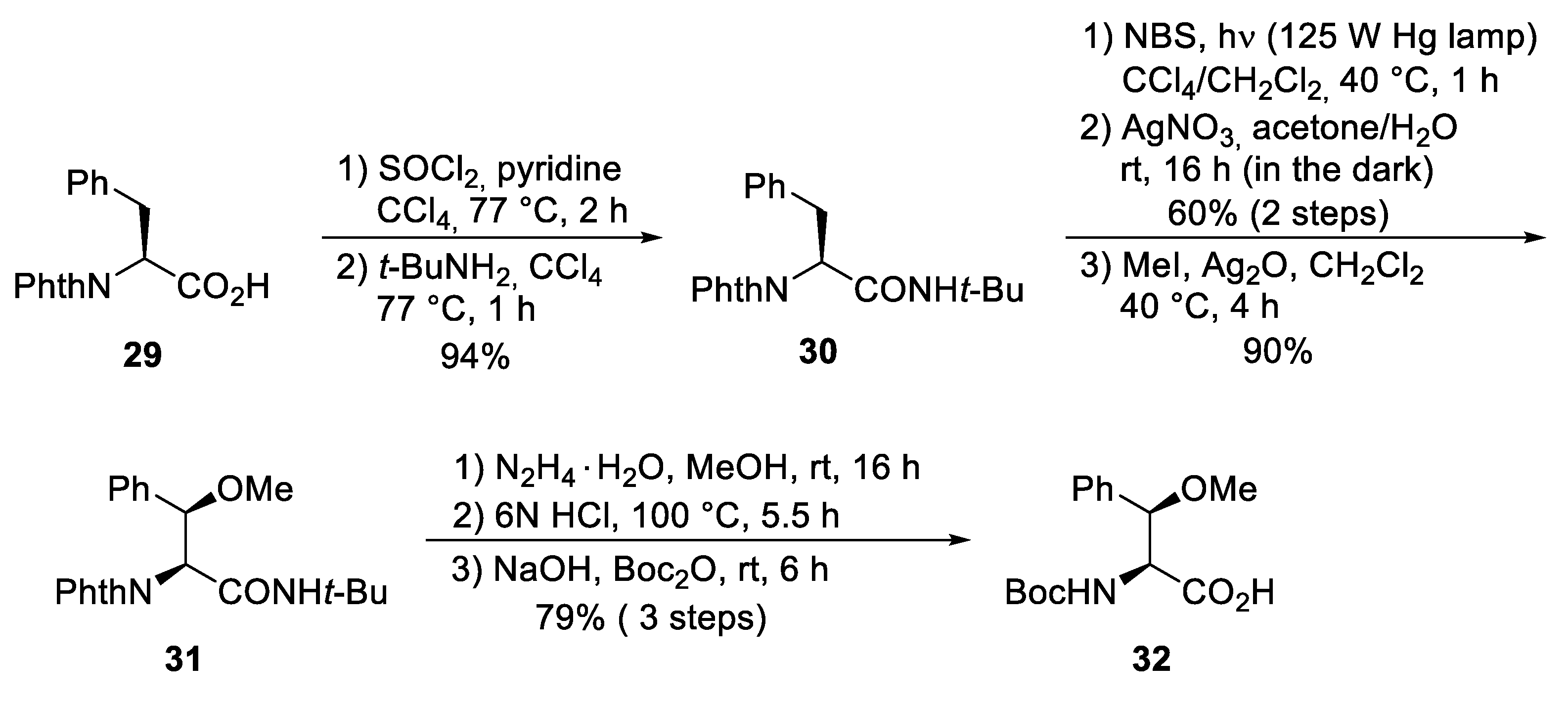
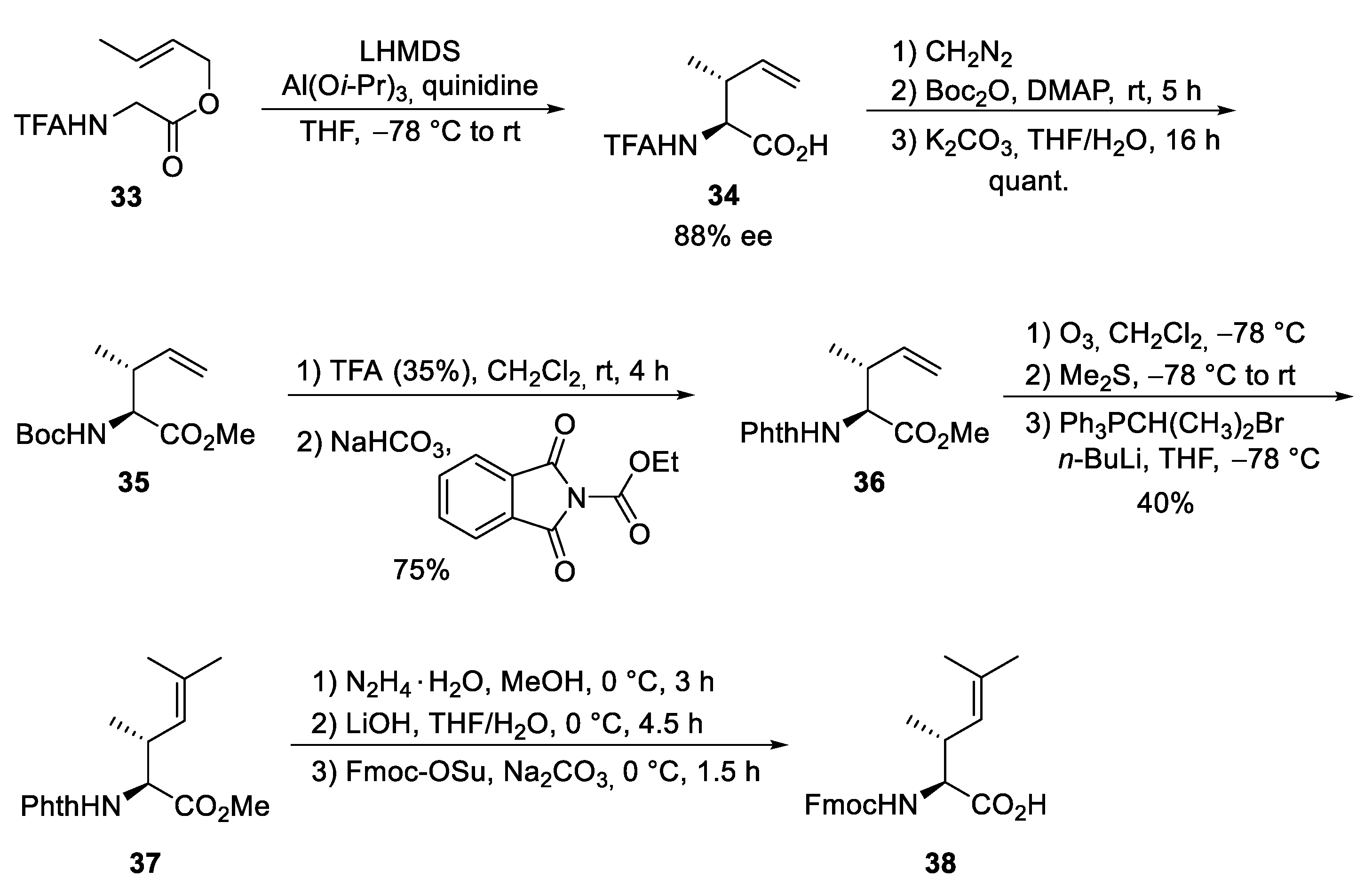
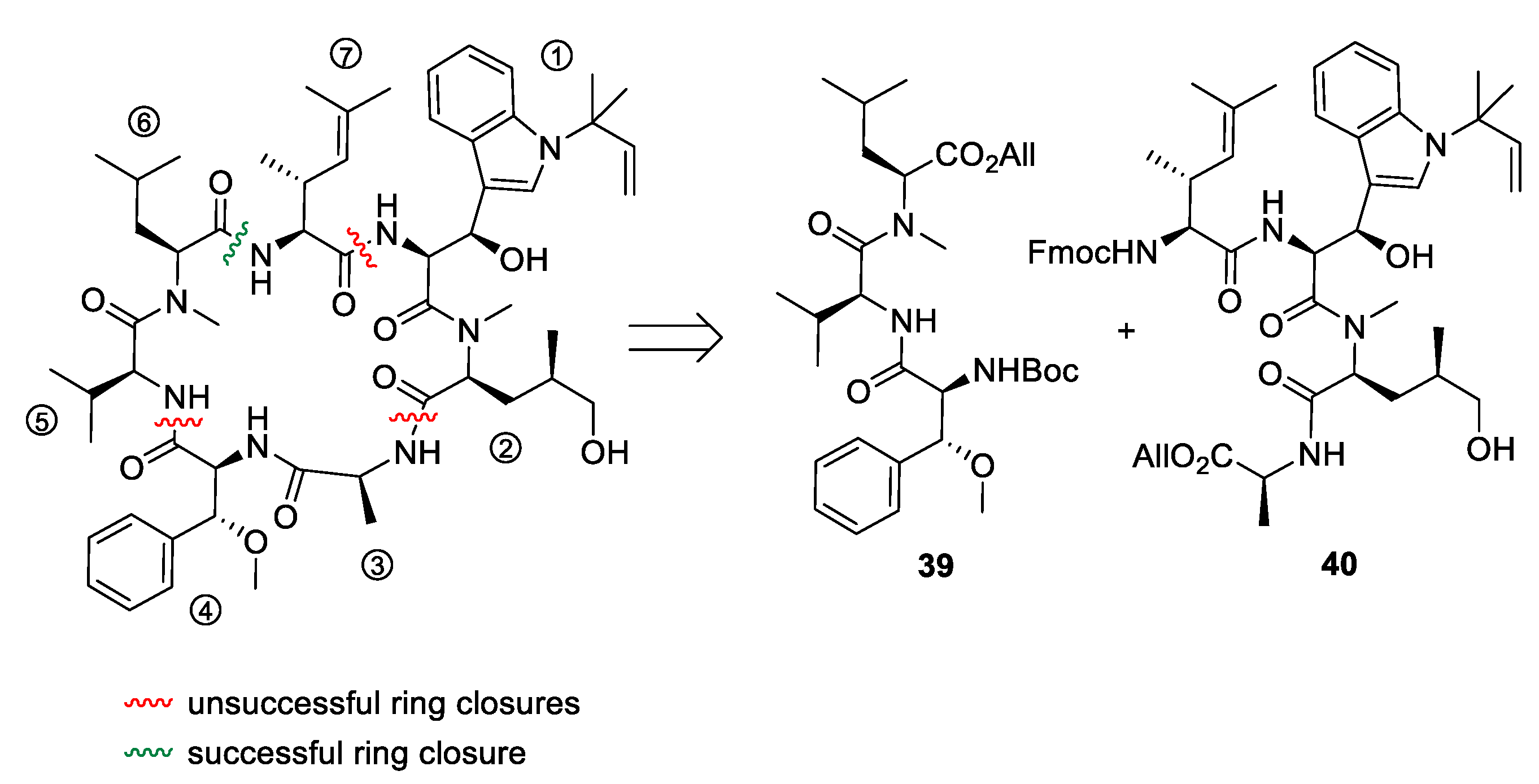
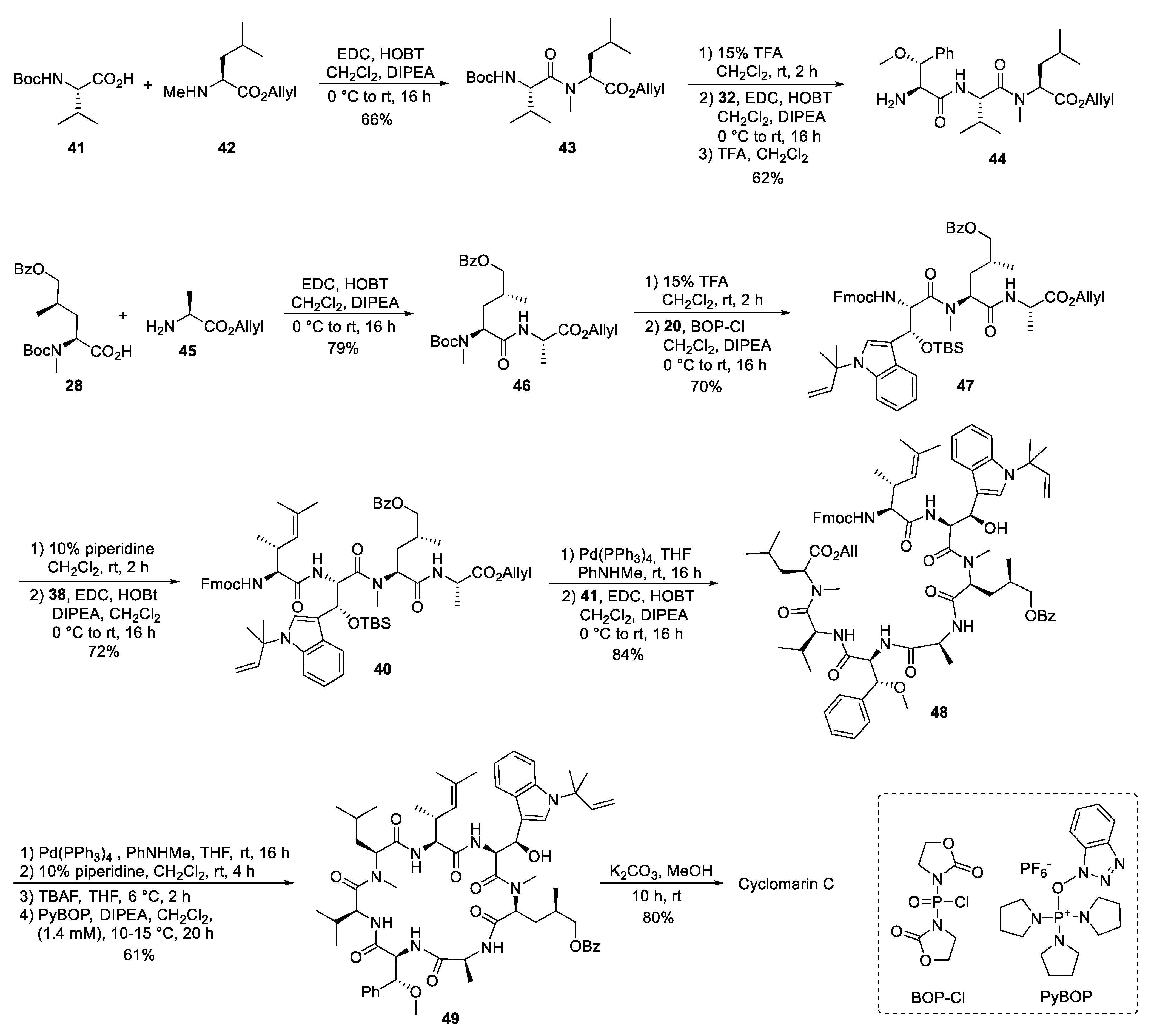
5.3. Total Synthesis of Cyclomarin Derivatives
6. Biological Activities and Mode of Action
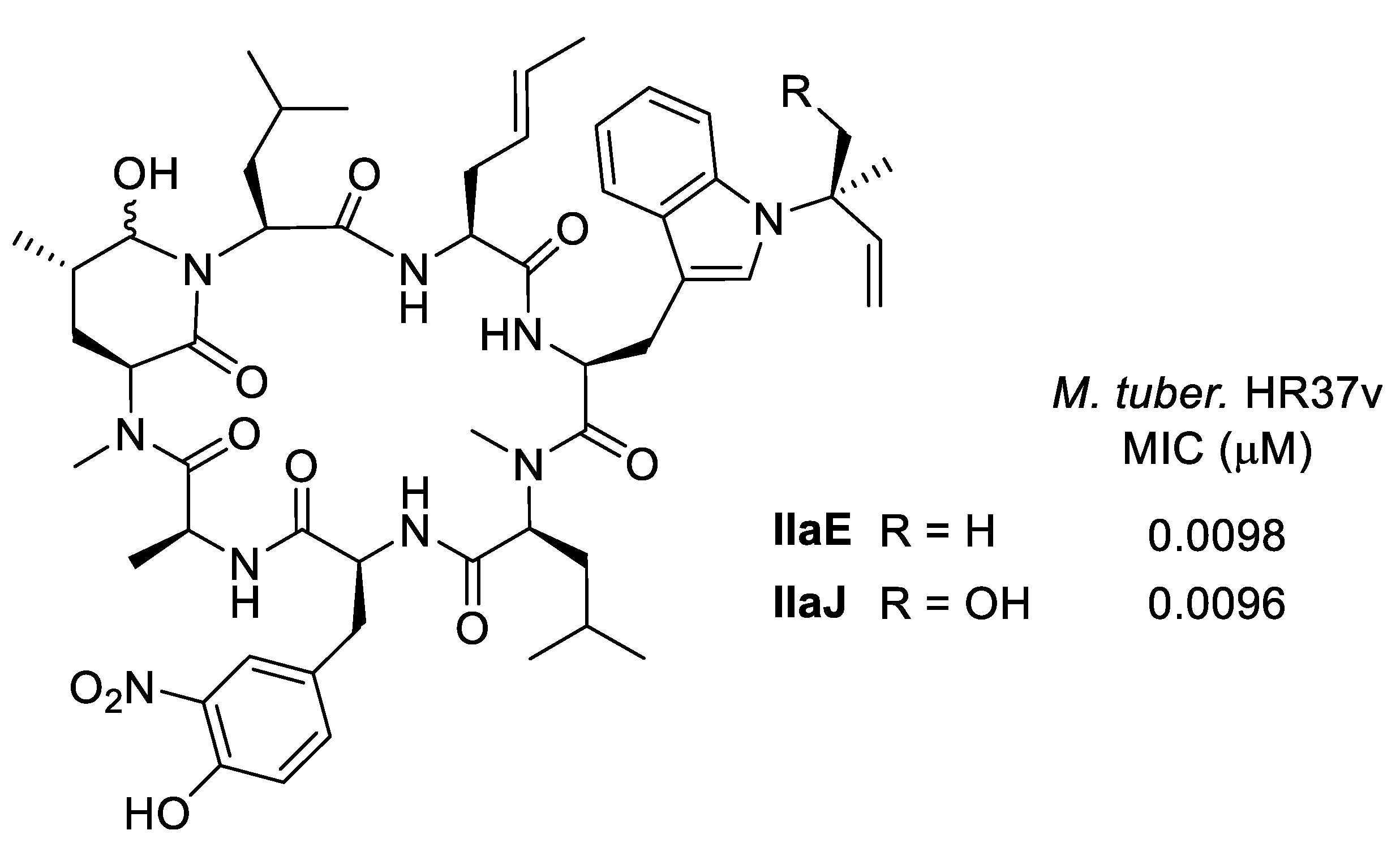
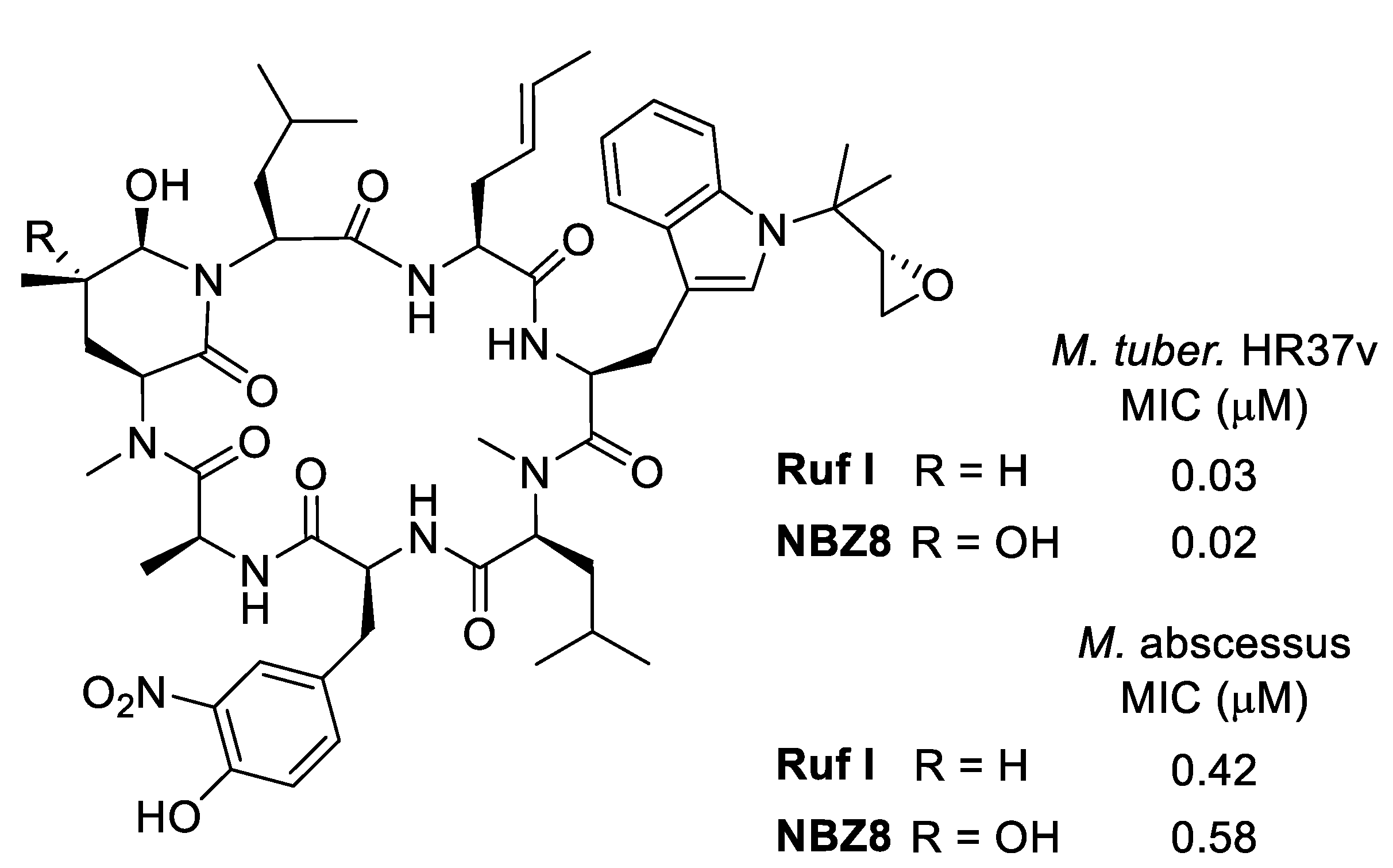
6.1. Biological Activities of Ilamycins/Rufomycins
6.2. Biological Activities of Cyclomarins
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Spane, V.; Montalbano, A.; Cueto, M.; Marrero, A.R.D.; Deniz, I.; Erdogan, A.; Bilela, L.L.; Moulin, C.; Taffin-de-Givenchy, E.; et al. Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes. Mar. Drugs 2020, 18, 619. [Google Scholar] [CrossRef] [PubMed]
- McCauley, E.P.; Pina, I.C.; Thompson, A.D.; Bashir, K.; Weinberg, M.; Kurz, S.L.; Crews, P. Highlights of marine natural products having parallel scaffolds found from marine-derived bacteria, sponges, and tunicates. J. Antibiot. 2020, 73, 504–525. [Google Scholar] [CrossRef] [PubMed]
- Qamar, H.; Hussain, K.; Soni, A.; Khan, A.; Hussain, T.; Chenais, B. Cyanobacteria as natural therapeutics and pharmaceutical potential: Role in antitumor activity and as nanovectors. Molecules 2021, 26, 247. [Google Scholar] [CrossRef] [PubMed]
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.B.; Smith, P.W.; Chibale, K. Recent approaches to chemical discovery and development against malaria and the neglected tropical diseases human african trypanosomiasis and schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/health-topics/tuberculosis#tab=tab_1 (accessed on 30 July 2021).
- De Opitz, C.L.M.; Sass, P. Tackling antimicrobial resistance by exploring new mechanisms of antibiotic action. Future Microbiol. 2020, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Dooley, K.E.; Park, J.G.; Swindells, S.; Allen, R.; Haas, D.W.; Cramer, Y.; Aweeka, F.; Wiggins, I.; Gupta, A.; Lizak, P.; et al. Safety, tolerability, and pharmacokinetic interactions of the antituberculous agent TMC207 (bedaquiline) with efavirenz in healthy volunteers: AIDS clinical trials group study A5267. J. Acq. Imm. Def. 2012, 59, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Igarashi, M.; Ishizaki, Y.; Takahashi, Y. New antituberculous drugs derived from natural products: Current perspectives and issues in antituberculous drug development. J. Antibiot. 2018, 71, 15–25. [Google Scholar] [CrossRef]
- Lee, H.; Suh, J.W. Anti-tuberculosis lead molecules from natural products targeting Mycobacterium tuberculosis ClpC1. J. Ind. Microbiol. Biotechnol. 2016, 43, 205–212. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Takita, T.; Ozawa, H.; Umezawa, H.; Tahara, K. Studies on ilamycin. J. Antibiot. 1962, 15, 49–50. [Google Scholar]
- Takita, T.; Ohi, K.; Maeda, K.; Okami, Y.; Umezawa, H. New antibiotics, ilamycins. J. Antibiot. 1962, 15, 46–48. [Google Scholar]
- Higashidani, E.; Ueyanagi, J.; Shibata, M.; Nakazawa, K.; Miyake, A.; Iwasaki, H.; Yamamoto, H. Studies on Streptomycetes. 2. Rufomycin A and A, new antituberculous antibiotics. Agric. Biol. Chem. 1962, 26, 234–237. [Google Scholar]
- Shibata, M.; Yamamoto, H.; Higashidani, E.; Nakazawa, K. Studies on Streptomycetes. 1. Streptromyces atratus nov. sp. producing new antituberculous antibiotics rufomycin A and B. Agric. Biol. Chem. 1962, 26, 228–233. [Google Scholar]
- Cary, L.W.; Takita, T.; Ohnishi, M. Study of secondary structure of ilamycin-B1 by 300 MHz proton magnetic resonance. FEBS Lett. 1971, 17, 145–148. [Google Scholar] [CrossRef]
- Iitaka, Y.; Nakamura, H.; Takada, K.; Takita, T. X-RAY study of ilamycin B1, a cyclic heptapeptide antibiotic. Acta Cryst. B 1974, 30, 2817–2825. [Google Scholar] [CrossRef]
- Iwasaki, H.; Witkop, B. New methods for nonenzymatic peptide cleavage. Electrolytic differential + solvolytic cleavage of antibiotic cyclopeptide rufymacin. J. Am. Chem. Soc. 1964, 86, 4698–4708. [Google Scholar] [CrossRef]
- Takita, T. Amino acid sequence of ilamycin and ilamycin B. J. Antibiot. 1963, 16, 211–212. [Google Scholar]
- Takita, T.; Maeda, K.; Naganawa, H.; Umezawa, H. Structures of ilamycin and ilamycin B2. J. Antibiot. 1964, 17, 129–131. [Google Scholar]
- Takita, T.; Naganawa, H.; Maeda, K.; Umezawa, H. The structural diffference among ilamycin, ilamycin C1 and ilamycin C2. J. Antibiot. 1965, 18, 135–136. [Google Scholar]
- Fujino, M.; Kamiya, T.; Miyake, A.; Iwasaki, H.; Ueyanagi, J. Tryptophan moiety of rufomycin homologs. Chem. Pharm. Bull. 1964, 12, 390–1392. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takita, T.; Naganawa, H.; Maeda, K.; Umezawa, H. Further studies on tryptophan parts of ilamycins. J. Antibiot. 1964, 17, 264–265. [Google Scholar]
- Takita, T.; Naganawa, H. L-2-Amino-4-hexenoic acid in ilamycins. J. Antibiot. 1963, 16, 246. [Google Scholar]
- Ma, J.Y.; Huang, H.B.; Xie, Y.C.; Liu, Z.Y.; Zhao, J.; Zhang, C.Y.; Jia, Y.X.; Zhang, Y.; Zhang, H.; Zhang, T.Y.; et al. Biosynthesis of ilamycins featuring unusual building blocks and engineered production of enhanced anti-tuberculosis agents. Nat. Commun. 2017, 8, 391. [Google Scholar] [CrossRef]
- Sun, C.L.; Liu, Z.Y.; Zhu, X.C.; Fan, Z.Y.; Huang, X.M.; Wu, Q.L.; Zheng, X.H.; Qin, X.J.; Zhang, T.Y.; Zhang, H.; et al. Antitubercular ilamycins from marine-derived Streptomyces atratus SCSIO ZH16 Δ ilaR. J. Nat. Prod. 2020, 83, 1646–1657. [Google Scholar] [CrossRef]
- Zhou, B.; Shetye, G.; Yu, Y.; Santarsiero, B.D.; Klein, L.L.; Abad-Zapatero, C.; Wolf, N.M.; Cheng, J.; Jin, Y.; Lee, H.; et al. Antimycobacterial rufomycin analogues from Streptomyces atratus strain MJM3502. J. Nat. Prod. 2020, 83, 657–667. [Google Scholar] [CrossRef]
- Renner, M.K.; Shen, Y.C.; Cheng, X.C.; Jensen, P.R.; Frankmoelle, W.; Kauffman, C.A.; Fenical, W.; Lobkovsky, E.; Clardy, J. Cyclomarins A-C, new antiinflammatory cyclic peptides produced by a marine bacterium (Streptomyces sp.). J. Am. Chem. Soc. 1999, 121, 11273–11276. [Google Scholar] [CrossRef]
- Kumamoto, T.; Koshino, H.; Watanabe, D.; Matsumoto, Y.; Aoyama, K.; Harada, K.; Ishikawa, T.; Mikami, Y. M10709, a new peptide antibiotic from clinically isolated Streptomyces sp. Heterocycles 2010, 80, 281–288. [Google Scholar]
- Li, L.; MacIntyre, L.W.; Ali, T.; Russo, R.; Koirala, B.; Hernandez, Y.; Brady, S.F. Biosynthetic interrogation of soil metagenomes reveals metamarin, an uncommon cyclomarin congener with activity against Mycobacterium tuberculosis. J. Nat. Prod. 2021, 84, 1056–1066. [Google Scholar] [CrossRef]
- Tomita, H.; Katsuyama, Y.; Minami, H.; Ohnishi, Y. Identification and characterization of a bacterial cytochrome P450 monooxygenase catalyzing the 3-nitration of tyrosine in rufomycin biosynthesis. J. Biol. Chem. 2017, 292, 15859–15869. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.W.; Lewis, C.A.; Luzung, M.R.; Baran, P.S.; Moore, B.S. Functional characterization of the cyclomarin/cyclomarazine prenyltransferase CymD directs the biosynthesis of unnatural cyclic peptides. J. Nat. Prod. 2010, 73, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.W.; Oh, D.C.; Carney, J.R.; Williamson, R.T.; Udwary, D.W.; Jensen, P.R.; Gould, S.J.; Fenical, W.; Moore, B.S. Biosynthesis and structures of cyclomarins and cyclomarazines, prenylated cyclic peptides of marine actinobacterial origin. J. Am. Chem. Soc. 2008, 130, 4507–4516. [Google Scholar] [CrossRef] [PubMed]
- He, J.Q.; Wei, X.; Yang, Z.J.; Li, Y.; Ju, J.H.; Ma, J.Y. Characterization of regulatory and transporter genes in the biosynthesis of anti-tuberculosis ilamycins and production in a heterologous host. Mar. Drugs 2020, 18, 216. [Google Scholar] [CrossRef]
- Kiefer, A.; Kazmaier, U. Syntheses of cyclomarins—Interesting marine natural products with distinct mode of action towards malaria and tuberculosis. Synthesis 2019, 51, 107–121. [Google Scholar] [CrossRef]
- Cheng, Y.Y.; Tang, S.B.; Guo, Y.; Ye, T. Total synthesis of anti-tuberculosis natural products ilamycins E-1 and F. Org. Lett. 2018, 20, 6166–6169. [Google Scholar] [CrossRef]
- Luzung, M.R.; Lewis, C.A.; Baran, P.S. Direct, chemoselective N-tert-prenylation of indoles by C-H functionalization. Angew. Chem. Int. Ed. 2009, 48, 7025–7029. [Google Scholar] [CrossRef]
- Arda, A.; Soengas, R.G.; Nieto, M.I.; Jimenez, C.; Rodriguez, J. Total synthesis of (-)-dysithiazolamide. Org. Lett. 2008, 10, 2175–2178. [Google Scholar] [CrossRef]
- Hanessian, S.; Margarita, R. 1,3-asymmetric induction in dianionic allylation reactions of amino acid derivatives-synthesis of functionally useful enantiopure glutamates, pipecolates and pyroglutamates. Tetrahedron Lett. 1998, 39, 5887–5890. [Google Scholar] [CrossRef]
- Padron, J.M.; Kokotos, G.; Martin, T.; Markidis, T.; Gibbons, W.A.; Martin, V.S. Enantiospecific synthesis of alpha-amino acid semialdehydes: A key step for the synthesis of unnatural unsaturated and saturated alpha-amino acids. Tetrahedron Asymmetry 1998, 9, 3381–3394. [Google Scholar] [CrossRef]
- Schöllkopf, U.; Groth, U.; Deng, C. Asymmetric synthesis via heterocyclic intermediates. 6. Enantioselective synthesis of (R)-amino acids using valine as a chiral agent. Angew. Chem. Int. Ed. Engl. 1981, 20, 798–799. [Google Scholar] [CrossRef]
- Albericio, F.; Cases, M.; Alsina, J.; Triolo, S.A.; Carpino, L.A.; Kates, S.A. On the use of PyAOP, a phosphonium salt derived from HOAt, in solid-phase peptide synthesis. Tetrahedron Lett. 1997, 38, 4853–4856. [Google Scholar] [CrossRef]
- Chen, S.Q.; Xu, J.C. Pentafluorophenyl diphenylphosphinate—A new efficient coupling reagent in peptide chemistry. Tetrahedron Lett. 1991, 32, 6711–6714. [Google Scholar] [CrossRef]
- Sathish, K.; Reddy, G.P.K.; Mainkar, P.S.; Chandrasekhar, S. Synthesis of the ‘southern’ tripeptide of Cyclomarins A and C having novel anti-tuberculocidal mode of action. Tetrahedron Asymmetry 2011, 22, 1568–1573. [Google Scholar] [CrossRef]
- Wen, S.J.; Yao, Z.J. Total synthesis of cyclomarin C. Org. Lett. 2004, 6, 2721–2724. [Google Scholar] [CrossRef]
- Wen, S.J.; Zhang, H.W.; Yao, Z.J. Synthesis of a fully protected (2S,3R)-N-(1′,1′-dimethyl-2′-propenyl)-3-hydroxytryptophan from tryptophan. Tetrahedron Lett. 2002, 43, 5291–5294. [Google Scholar] [CrossRef]
- Tao, B.; Schlingloff, G.; Sharpless, K.B. Reversal of regioselection in the asymmetric aminohydroxylation of cinnamates. Tetrahedron Lett. 1998, 39, 2507–2510. [Google Scholar] [CrossRef]
- Barbie, P.; Kazmaier, U. Synthesis of fully protected, reverse N-prenylated (2S,3R)-3-hydroxytryptophan, a unique building block of the cyclomarins. Org. Biomol. Chem. 2015, 13, 9267–9275. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Evans, D.A.; Ripin, D.H.B.; Halstead, D.P.; Campos, K.R. Synthesis and absolute stereochemical assignment of (+)-miyakolide. J. Am. Chem. Soc. 1999, 121, 6816–6826. [Google Scholar] [CrossRef]
- Easton, C.J.; Hutton, C.A.; Roselt, P.D.; Tiekink, E.R.T. Synthesis and molecular structure of stable derivatives of (E)-dehydrophenylalanine and (Z)-dehydrophenylalanine. Aust. J. Chem. 1991, 44, 687–694. [Google Scholar] [CrossRef]
- Easton, C.J.; Hutton, C.A.; Roselt, P.D.; Tiekink, E.R.T. Stereocontrolled synthesis of β-hydroxyphenylalanine and β-hydroxytyrosine derivatives. Tetrahedron 1994, 50, 7327–7340. [Google Scholar] [CrossRef]
- Kazmaier, U.; Krebs, A. Synthesis of chiral γ,δ-unsaturated amino acids by asymmetric ester enolate Claisen rearrangement. Angew. Chem. Int. Ed. Engl. 1995, 34, 2012–2014. [Google Scholar] [CrossRef]
- Kazmaier, U.; Mues, H.; Krebs, A. Asymmetric chelated Claisen rearrangements in the presence of chiral ligands—Scope and limitations. Chem. Eur. J. 2002, 8, 1850–1855. [Google Scholar] [CrossRef]
- Wen, S.J.; Hu, T.S.; Yao, Z.J. Macrocyclization studies and total synthesis of cyclomarin C, an anti-inflammatory marine cyclopeptide. Tetrahedron 2005, 61, 4931–4938. [Google Scholar] [CrossRef]
- Cabre, J.; Palomo, A.L. New experimental strategies in amide synthesis using N,N-Bis-2-oxo-3-oxazolidinyl phosphordiamidic chloride. Synthesis 1984, 413–417. [Google Scholar] [CrossRef]
- Coste, J.; Lenguyen, D.; Castro, B. PYBOP—A new peptide coupling reagent devoid of toxic byproducts. Tetrahedron Lett. 1990, 31, 205–208. [Google Scholar] [CrossRef]
- Barbie, P.; Kazmaier, U. Total Synthesis of Cyclomarin A, a Marine Cycloheptapeptide with Anti-Tuberculosis and Anti-Malaria Activity. Org. Lett. 2016, 18, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.F.; Van Zeeland, R.; Stanley, L.M. Palladium-catalyzed synthesis of N-tert-prenylindoles. Org. Lett. 2013, 15, 2798–2801. [Google Scholar] [CrossRef]
- Sugiyama, H.; Shioiri, T.; Yokokawa, F. Syntheses of four unusual amino acids, constituents of cyclomarin A. Tetrahedron Lett. 2002, 43, 3489–3492. [Google Scholar] [CrossRef]
- Della Sala, G.; Izzo, I.; Spinella, A. A Pd-mediated approach to the synthesis of an unusual β-hydroxytryptophan amino acid constituent of cyclomarin A. Synlett 2006, 1319–1322. [Google Scholar]
- Metzger, A.; Bernhardt, S.; Manolikakes, G.; Knochel, P. MgCl2-accelerated addition of functionalized organozinc reagents to aldehydes, ketones, and carbon dioxide. Angew. Chem. Int. Ed. 2010, 49, 4665–4668. [Google Scholar] [CrossRef] [PubMed]
- Piller, F.M.; Metzger, A.; Schade, M.A.; Haag, B.A.; Gavryushin, A.; Knochel, P. Preparation of polyfunctional arylmagnesium, arylzinc, and benzylic zinc reagents by using magnesium in the presence of LiCl. Chem. Eur. J. 2009, 15, 7192–7202. [Google Scholar] [CrossRef]
- Corey, E.J.; Venkateswarlu, A. Protection oh hydroxyl groups as tert-Butyldimethylsilyl derivatives. J. Am. Chem. Soc. 1972, 94, 6190–6191. [Google Scholar] [CrossRef]
- Futagawa, S.; Inui, T.; Shiba, T. Nuclear magnetic-resonance study of stereoisomeric 2-oxazolidone and 2-phenyl-2-oxazoline derivatives of α-amino-β-hydroxy acids. Bull. Chem. Soc. Jpn. 1973, 46, 3308–3310. [Google Scholar] [CrossRef]
- Parikh, J.R.; Doering, W.V.E. Sulfur trioxide in oxidation of alcohols by dimethyl sulfoxide. J. Am. Chem. Soc. 1967, 89, 5505–5507. [Google Scholar] [CrossRef]
- McDonald, C.; Holcomb, H.; Kennedy, K.; Kirkpatrick, E.; Leathers, T.; Vanemon, P. N-iodosuccinimide-mediated conversion of aldehydes to methyl esters. J. Org. Chem. 1989, 54, 1213–1215. [Google Scholar] [CrossRef]
- Schmidt, U.; Griesser, H.; Leitenberger, V.; Lieberknecht, A.; Mangold, R.; Meyer, R.; Riedl, B. Amino-acids and peptides. 81. Diastereoselective formation of (Z)-didehydroamino acid-esters. Synthesis 1992, 487–490. [Google Scholar] [CrossRef]
- Schmidt, U.; Lieberknecht, A.; Kazmaier, U.; Griesser, H.; Jung, G.; Metzger, J. Amino-acids and peptides. 75. Synthesis of dihydroxyamino and trihydroxyamino acids—Construction of lipophilic tripalmitoyldihydroxy-α-amino acids. Synthesis 1991, 49–55. [Google Scholar] [CrossRef]
- Panella, L.; Aleixandre, A.M.; Kruidhof, G.J.; Robertus, J.; Feringa, B.L.; de Vries, J.G.; Minnaard, A.J. Enantioselective Rh-catalyzed hydrogenation of N-formyl dehydroamino esters with monodentate phosphoramidite ligands. J. Org. Chem. 2006, 71, 2026–2036. [Google Scholar] [CrossRef]
- Van den Berg, M.; Minnaard, A.J.; Schudde, E.P.; van Esch, J.; de Vries, A.H.M.; de Vries, J.G.; Feringa, B.L. Highly enantioselective rhodium-catalyzed hydrogenation with monodentate ligands. J. Am. Chem. Soc. 2000, 122, 11539–11540. [Google Scholar] [CrossRef]
- Kiefer, A.; Kazmaier, U. Synthesis of modified β-methoxyphenylalanines via diazonium chemistry and their incorporation in desoxycyclomarin analogues. Org. Biomol. Chem. 2019, 17, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Kazmaier, U.; Schneider, C. Stereoselective synthesis of unsaturated polyhydroxylated amino acids via ester enolate Claisen rearrangement. Synlett 1996, 10, 975–977. [Google Scholar] [CrossRef]
- Kazmaier, U.; Schneider, C. Application of the asymmetric chelate enolate Claisen rearrangement to the synthesis of unsaturated polyhydroxylated amino acids. Synthesis 1998, 1321–1326. [Google Scholar] [CrossRef]
- Kazmaier, U.; Schneider, C. Application of the asymmetric chelate-enolate Claisen rearrangement to the synthesis of 5-epi-isofagomine. Tetrahedron Lett. 1998, 39, 817–818. [Google Scholar] [CrossRef]
- Marti, C.; Carreira, E.M. Total synthesis of (-)-spirotryprostatin B: Synthesis and related studies. J. Am. Chem. Soc. 2005, 127, 11505–11515. [Google Scholar] [CrossRef]
- Li, P.; Xu, J.C. 1-Ethyl 2-halopyridinium salts, highly efficient coupling reagents for hindered peptide synthesis both in solution and the solid-phase. Tetrahedron 2000, 56, 8119–8131. [Google Scholar] [CrossRef]
- Li, P.; Xu, J.C. 2-Bromo-1-ethyl pyridinium tetrafluoroborate (BEP): A powerful coupling reagent for N-methylated peptide synthesis. Chem. Lett. 2000, 29, 204–205. [Google Scholar] [CrossRef]
- Barbie, P.; Kazmaier, U. Total synthesis of cyclomarins A, C and D, marine cyclic peptides with interesting anti-tuberculosis and anti-malaria activities. Org. Biomol. Chem. 2016, 14, 6036–6054. [Google Scholar] [CrossRef]
- Junk, L.; Papadopoulos, E.; Kazmaier, U. Tryptophan N-1-alkylation: Quick and simple access to diversely substituted tryptophans. Synthesis 2021, 53, 2503–2511. [Google Scholar] [CrossRef]
- Bürstner, N.; Roggo, S.; Ostermann, N.; Blank, J.; Delmas, C.; Freuler, F.; Gerhartz, B.; Hinniger, A.; Hoepfner, D.; Liechty, B.; et al. Gift from Nature: Cyclomarin A Kills Mycobacteria and Malaria Parasites by Distinct Modes of Action. ChemBioChem 2015, 16, 2433–2436. [Google Scholar] [CrossRef]
- Schmitt, E.K.; Riwanto, M.; Sambandamurthy, V.; Roggo, S.; Miault, C.; Zwingelstein, C.; Krastel, P.; Noble, C.; Beer, D.; Rao, S.P.S.; et al. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem. Int. Ed. 2011, 50, 5889–5891. [Google Scholar] [CrossRef]
- Vasudevan, D.; Rao, S.P.S.; Noble, C.G. Structural basis of mycobacterial inhibition by cyclomarin A. J. Biol. Chem. 2013, 288, 30883–30891. [Google Scholar] [CrossRef]
- Barbie, P.; Kazmaier, U. Total synthesis of desoxycyclomarin C and the cyclomarazines A and B. Org. Biomol. Chem. 2016, 14, 6055–6064. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, A.; Bader, C.D.; Held, J.; Esser, A.; Rybniker, J.; Empting, M.; Müller, R.; Kazmaier, U. Synthesis of new cyclomarin derivatives and their biological evaluation towards Mycobacterium tuberculosis and plasmodium falciparum. Chem. Eur. J. 2019, 25, 8894–8902. [Google Scholar] [CrossRef] [PubMed]
- Choules, M.P.; Wolf, N.M.; Lee, H.; Anderson, J.R.; Grzelak, E.M.; Wang, Y.H.; Ma, R.; Gao, W.; McAlpine, J.B.; Jin, Y.Y.; et al. Rufomycin targets ClpC1 proteolysis in Mycobacterium tuberculosis and M. abscessus. Antimicrob. Agents Chemother. 2019, 63, e02204-18. [Google Scholar] [CrossRef] [PubMed]
- Nessar, R.; Cambau, E.; Reyrat, J.M.; Murray, A.; Gicquel, B. Mycobacterium abscessus: A new antibiotic nightmare. J. Antimicrob. Chemother. 2012, 67, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Kar, N.P.; Sikriwal, D.; Rath, P.; Choudhary, R.K.; Batra, J.K. Mycobacterium tuberculosis ClpC1. FEBS J. 2008, 275, 6149–6158. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Sauer, R.T. Substrate delivery by the AAA+ ClpX and ClpC1 unfoldases activates the mycobacterial ClpP1P2 peptidase. Mol. Microbiol. 2014, 93, 617–628. [Google Scholar] [CrossRef]
- Wolf, N.M.; Lee, H.; Choules, M.P.; Pauli, G.F.; Phansalkar, R.; Anderson, J.R.; Gao, W.; Ren, J.H.; Santarsiero, B.D.; Lee, H.; et al. High-resolution structure of CIpC1-rufomycin and ligand binding studies provide a framework to design and optimize anti-tuberculosis leads. ACS Infect. Dis. 2019, 5, 829–840. [Google Scholar] [CrossRef]
- Wolf, N.; Lee, H.; Choules, M.; Klein, L.; Petukhova, V.; Tufano, M.; Phansalkar, R.; Gao, W.; Santarsiero, B.; Lee, H.; et al. High-resolution structure of Clpc1-Ntd-Rufomycin complex provides three-dimensional framework for optimization of cyclopeptide anti-Tb drug leads. Protein Sci. 2018, 27, 119–120. [Google Scholar]
- Wolf, N.M.; Lee, H.; Nam, J.; Hong, J.; Duc, N.M.; Ho, N.A.; Lee, H.; Suh, J.W.; Pauli, G.F.; Franzblau, S.G.; et al. Structures of CIpC1-NTD with potent anti-TB cyclic peptides Rufomycin and Ecumicin: Implications for the mechanism of action and design of therapeutic agents. Acta Cryst. A 2019, 75, A59. [Google Scholar] [CrossRef]
- Xie, Q.; Yang, Z.J.; Huang, X.M.; Zhang, Z.K.; Li, J.B.; Ju, J.H.; Zhang, H.; Ma, J.Y. Ilamycin C induces apoptosis and inhibits migration and invasion in triple-negative breast cancer by suppressing IL-6/STAT3 pathway. J. Hematol. Oncol. 2019, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Intaraudom, C.; Rachtawee, P.; Suvannakad, R.; Pittayakhajonwut, P. Antimalarial and antituberculosis substances from Streptomyces sp. BCC26924. Tetrahedron 2011, 67, 7593–7597. [Google Scholar] [CrossRef]
- Weinhäupl, K.; Brennich, M.; Kazmaier, U.; Lelievre, J.; Ballell, L.; Goldberg, A.; Schanda, P.; Fraga, H. The antibiotic cyclomarin blocks arginine-phosphate-induced millisecond dynamics in the N-terminal domain of ClpC1 from Mycobacterium tuberculosis. J. Biol. Chem. 2018, 293, 8379–8393. [Google Scholar] [CrossRef]
- Maurer, M.; Linder, D.; Franke, K.B.; Jager, J.; Taylor, G.; Gloge, F.; Gremer, S.; Le Breton, L.; Mayer, M.P.; Weber-Ban, E.; et al. Toxic activation of an AAA plus protease by the antibacterial drug cyclomarin A. Cell Chem. Biol. 2019, 26, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | Mtb | Pfalcp | |
| wt Erdman MIC[µM] | 3D7 IC50[nM] | Dd2 IC50[nM] | |||||
| Isoniazid | 0.9 | ||||||
| CQ | 3.4 | 233.9 | |||||
| Cyclomarin A |  |  | Me | H | 0.125 | 36.8 | 27.7 |
| Cyclomarin C |  |  | Me | H | 0.25 | 42.8 | |
| Desoxycyclomarin C |  |  | Me | H | 0.9 | 39.8 | |
| 87a |  |  | Me | H | 0.5 | 9.0 | 12.9 |
| 87b |  |  | Me | H | 1.7 | 4.4 | 6.5 |
| 87c | iPr |  | Me | H | 0.4 | 47.8 | 76.0 |
| 87d | Me |  | Me | H | 0.9 | 13.4 | 17.8 |
| 87e | H |  | Me | H | 1.5 | 28.1 | 27.5 |
| 88a | iPr |  | Me | H | 0.4 | 355.7 | 300.3 |
| 88b | Me |  | Me | H | 1.6 | 230.3 | 256.7 |
| 88c | H |  | Me | H | 0.5 | 314.2 | 421.5 |
| 89a |  |  | H | H | 2.3 | 47.9 | 36.7 |
| 89b | iPr |  | H | H | 1.8 | 177.5 | 287.8 |
| 89c | Me |  | H | H | 3.4 | 362.4 | 318.4 |
| 90a |  |  | Me | NH2 | 0.25 | ||
| 90b |  |  | Me | N3 | 0.26 | ||
| 90c |  |  | Me | NO2 | 0.13 | ||
| 90d |  |  | Me | Br | 4.1 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kazmaier, U.; Junk, L. Recent Developments on the Synthesis and Bioactivity of Ilamycins/Rufomycins and Cyclomarins, Marine Cyclopeptides That Demonstrate Anti-Malaria and Anti-Tuberculosis Activity. Mar. Drugs 2021, 19, 446. https://doi.org/10.3390/md19080446
Kazmaier U, Junk L. Recent Developments on the Synthesis and Bioactivity of Ilamycins/Rufomycins and Cyclomarins, Marine Cyclopeptides That Demonstrate Anti-Malaria and Anti-Tuberculosis Activity. Marine Drugs. 2021; 19(8):446. https://doi.org/10.3390/md19080446
Chicago/Turabian StyleKazmaier, Uli, and Lukas Junk. 2021. "Recent Developments on the Synthesis and Bioactivity of Ilamycins/Rufomycins and Cyclomarins, Marine Cyclopeptides That Demonstrate Anti-Malaria and Anti-Tuberculosis Activity" Marine Drugs 19, no. 8: 446. https://doi.org/10.3390/md19080446
APA StyleKazmaier, U., & Junk, L. (2021). Recent Developments on the Synthesis and Bioactivity of Ilamycins/Rufomycins and Cyclomarins, Marine Cyclopeptides That Demonstrate Anti-Malaria and Anti-Tuberculosis Activity. Marine Drugs, 19(8), 446. https://doi.org/10.3390/md19080446