
New Ceramides and Cerebrosides from the Deep-Sea Far Eastern Starfish Ceramaster patagonicus

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
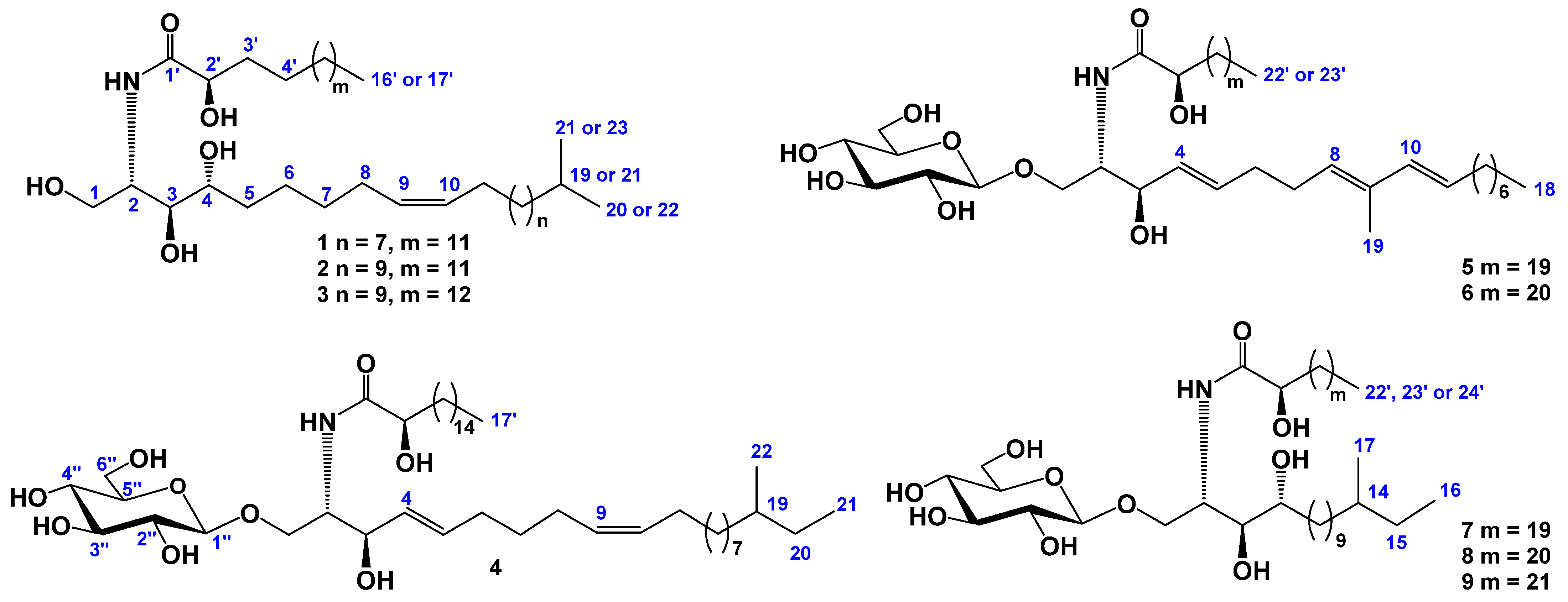
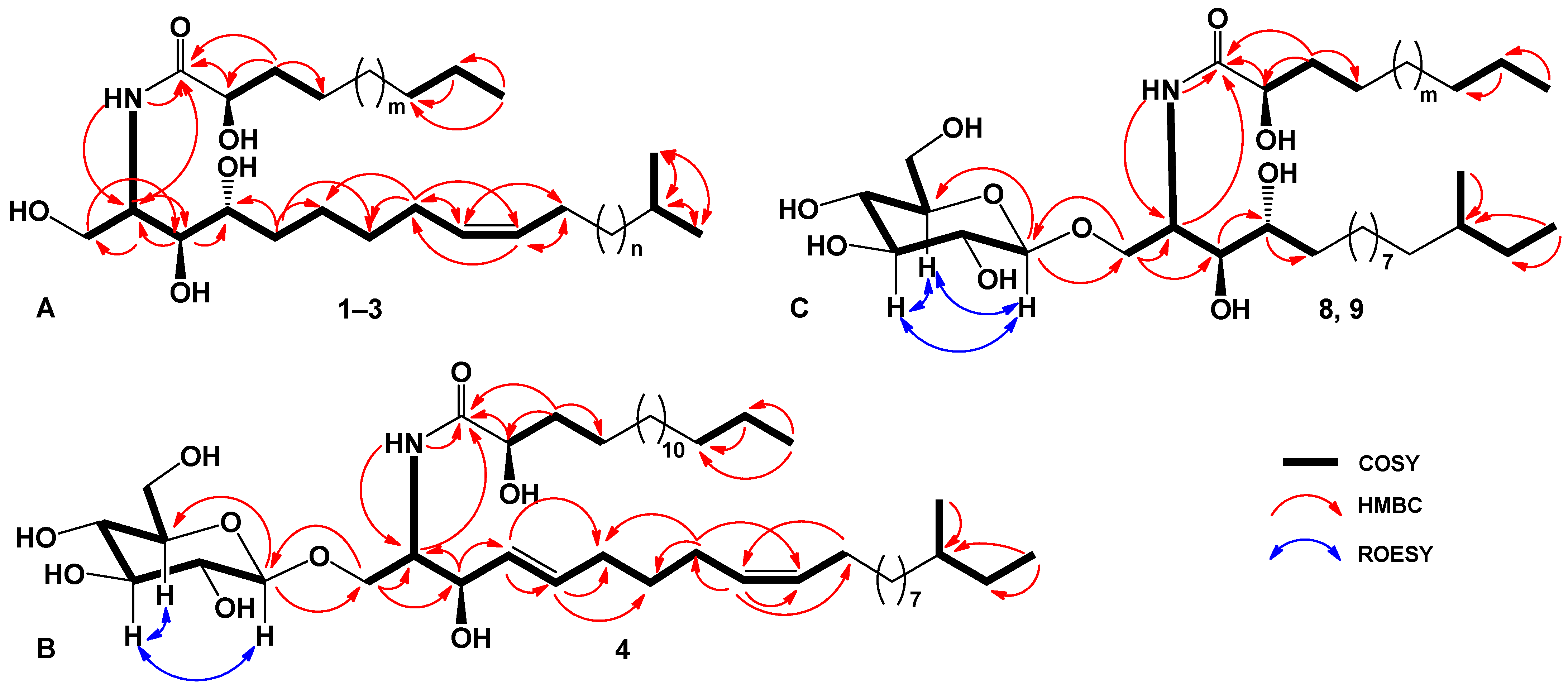
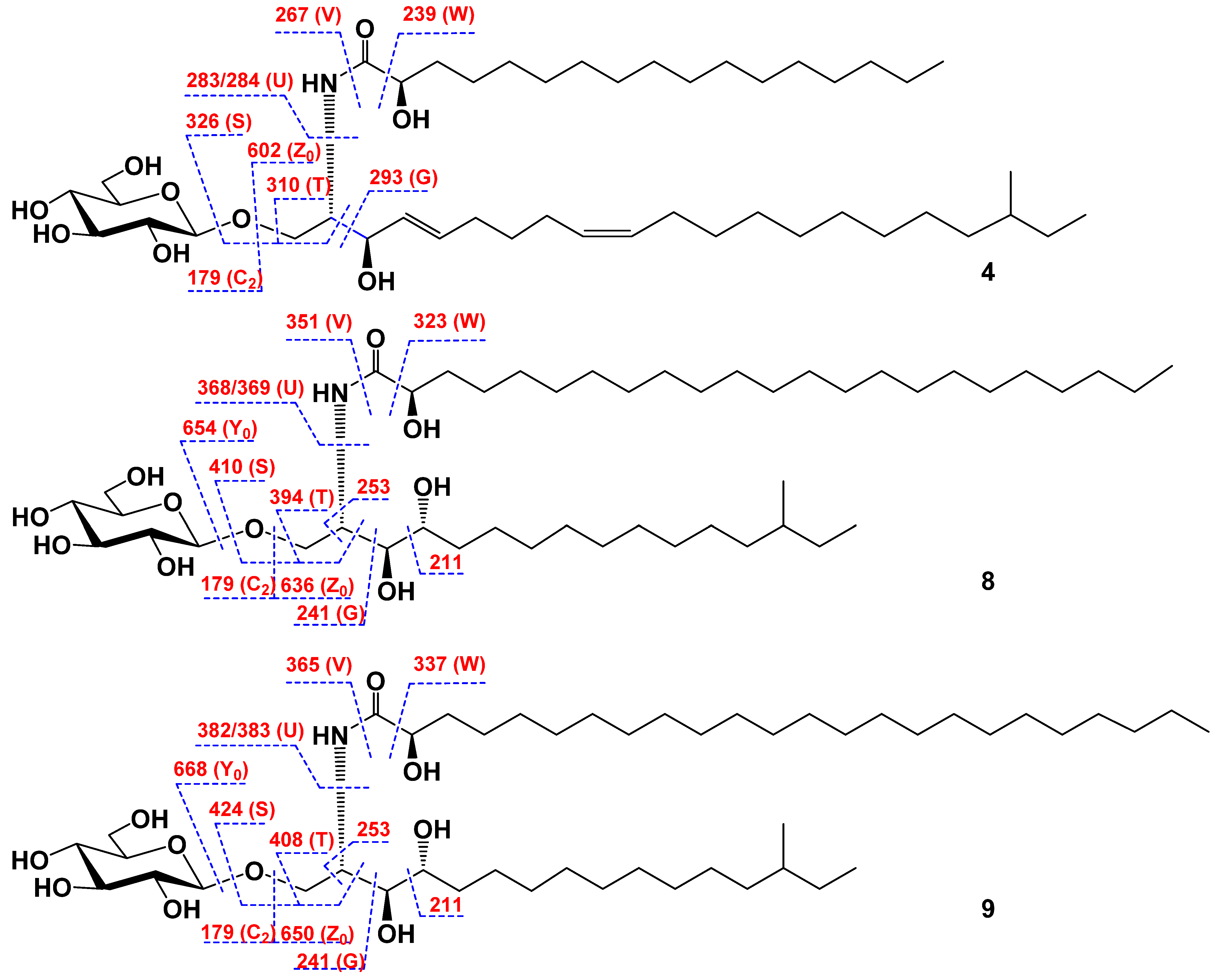
2.1. The Isolation and Determination of the Structures of Compounds 1–9 from C. patagonicus
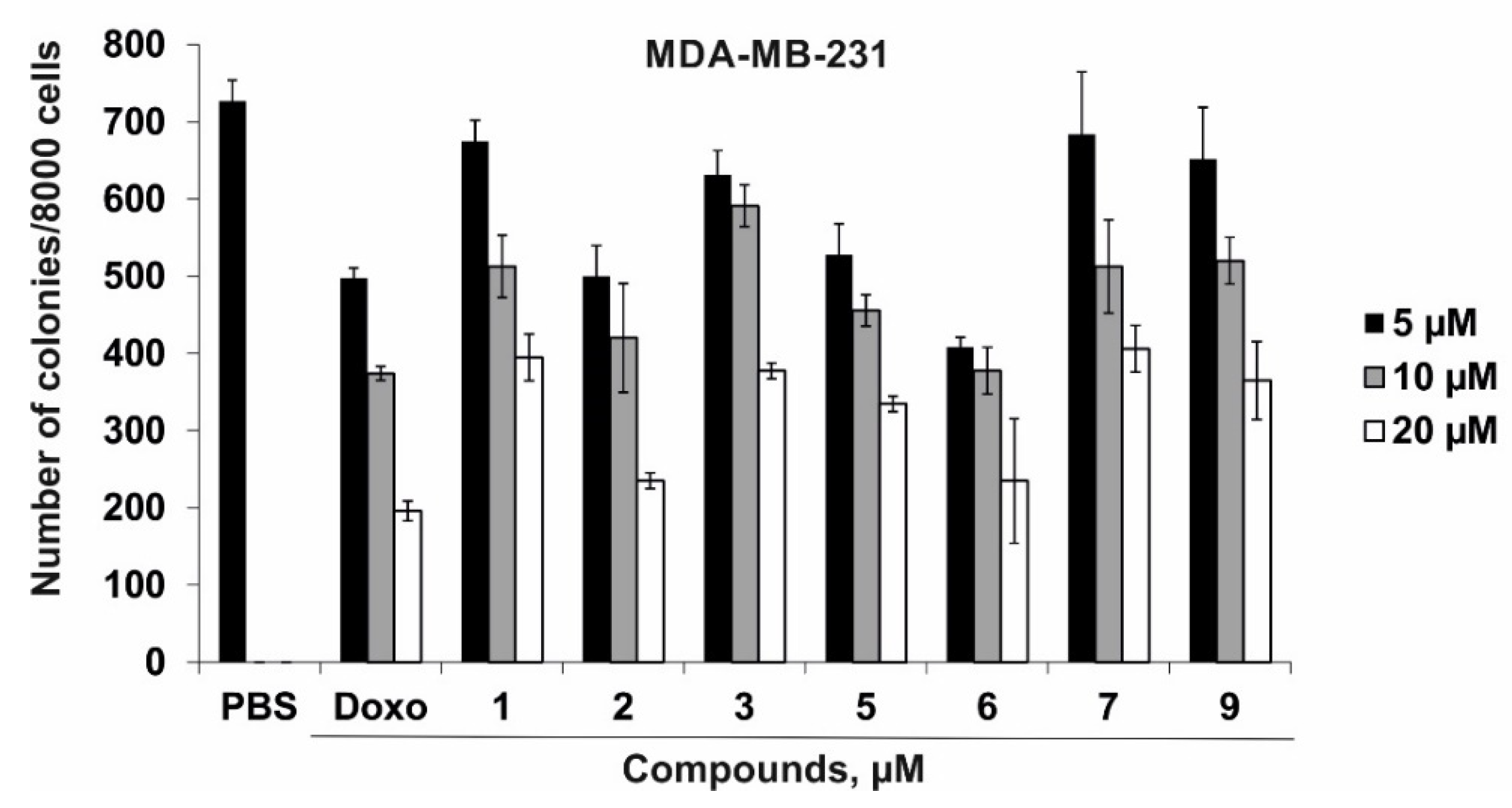
2.2. The Cytotoxic Activity of Compounds 1–3, 5–7, and 9 against Normal and Cancer Cells and their Effect on Colony Formation and Growth of Human Cancer Cells
3. Materials and Methods
3.1. General Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Compounds Characterization Data
3.5. Methanolysis of Compounds 1–9 and Analysis of FAMEs
3.6. Acid Hydrolysis and Determination of Absolute Configurations of Monosaccharides
3.7. Bioactivity Assay
3.7.1. Reagents
3.7.2. Cell Lines
3.7.3. Cells Culture Conditions
3.7.4. Preparation of Compounds
3.7.5. Cell Viability Assay
3.7.6. Colony Formation Assay
3.7.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Minale, L.; Riccio, R.; Zollo, F. Steroidal oligoglycosides and polyhydroxysteroids from Echinoderms. Fortschr. Chem. Org. Nat. 1993, 62, 75–308. [Google Scholar] [CrossRef]
- Stonik, V.A. Marine polar steroids. Russ. Chem. Rev. 2001, 70, 673–715. [Google Scholar] [CrossRef]
- Iorizzi, M.; De Marino, S.; Zollo, F. Steroidal oligoglycosides from the Asteroidea. Curr. Org. Chem. 2001, 5, 951–973. [Google Scholar] [CrossRef]
- Stonik, V.A.; Ivanchina, N.V.; Kicha, A.A. New polar steroids from starfish. Nat. Prod. Commun. 2008, 3, 1587–1610. [Google Scholar] [CrossRef]
- Dong, G.; Xu, T.H.; Yang, B.; Lin, X.P.; Zhou, X.F.; Yang, X.W.; Liu, Y.H. Chemical constituents and bioactivities of starfish. Chem. Biodivers. 2011, 8, 740–791. [Google Scholar] [CrossRef]
- Ivanchina, N.V.; Kicha, A.A.; Stonik, V.A. Steroid glycosides from marine organisms. Steroids 2011, 76, 425–454. [Google Scholar] [CrossRef] [PubMed]
- Ivanchina, N.V.; Kicha, A.A.; Malyarenko, T.V.; Stonik, V.A. Advances in Natural Products Discovery; Gomes, A.R., Rocha-Santos, T., Duarte, A., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2017; Volume 6, pp. 191–224. [Google Scholar]
- Stonik, V.A.; Kicha, A.A.; Malyarenko, T.V.; Ivanchina, N.V. Asterosaponins: Structures, taxonomic distribution, biogenesis and biological activities. Mar. Drugs 2020, 18, 584. [Google Scholar] [CrossRef]
- Xia, J.M.; Miao, Z.; Xie, C.L.; Zhang, J.W.; Yang, X.W. Chemical constituents and bioactivities of starfish: An update. Chem. Biodivers. 2020, 17, e1900638. [Google Scholar] [CrossRef] [PubMed]
- Muralidhar, P.; Radhika, P.; Krishna, N.; Rao, D.V.; Rao, C.B. Sphingolipids from marine organisms: A review. Nat. Prod. Sci. 2003, 9, 117–142. [Google Scholar]
- Higuchi, R.; Inagaki, M.; Yamada, K.; Miyamoto, T. Biologically active gangliosides from echinoderms. J. Nat. Med. 2007, 61, 367–370. [Google Scholar] [CrossRef]
- Yamada, K. Chemo-pharmaceutical studies on the glycosphingolipid constituents from Echinoderm, sea cucumbers, as the medicinal materials. Yakugaku Zasshi 2002, 122, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, M. Structure and biological activity of glycosphingolipids from starfish and feather star. Yakugaku Zasshi 2008, 128, 1187–1194. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Malyarenko, T.V.; Kicha, A.A.; Stonik, V.A.; Ivanchina, N.V. Sphingolipids of Asteroidea and Holothuroidea: Structures and biological activities. Mar. Drugs 2021, 19, 330. [Google Scholar] [CrossRef]
- Degroote, S.; Wolthoorn, J.; van Meer, G. The cell biology of glycosphingolipids. Semin. Cell Dev. Biol. 2004, 15, 375–387. [Google Scholar] [CrossRef]
- Hakomori, S.; Igarashi, Y. Functional role of glycosphingolipids in cell recognition and signaling. J. Biochem. 1995, 118, 1091–1103. [Google Scholar] [CrossRef]
- Sperling, P.; Heinz, E. Plant sphingolipids: Structural diversity, biosynthesis, first genes and functions. Biochem. Biophys. Acta 2003, 1632, 1–15. [Google Scholar] [CrossRef]
- Ishii, T.; Okino, T.; Mino, Y. A ceramide and cerebroside from the starfish Asterias amurensis Lütken and their plant-growth promotion activities. J. Nat. Prod. 2006, 69, 1080–1082. [Google Scholar] [CrossRef]
- Fischer, C.L.; Drake, D.R.; Dawson, D.V.; Blanchette, D.R.; Brogden, K.A.; Wertz, P.W. Antibacterial activity of sphingoid bases and fatty acids against gram-positive and gram-negative bacteria. Antimicrob. Agents Chemother. 2012, 56, 1157–1161. [Google Scholar] [CrossRef]
- Malyarenko, T.V.; Kicha, A.A.; Malyarenko, O.S.; Zakharenko, V.M.; Kotlyarov, I.P.; Kalinovsky, A.I.; Popov, R.S.; Svetashev, V.I.; Ivanchina, N.V. New conjugates of polyhydroxysteroids with long-chain fatty acids from the deep-water Far Eastern starfish Ceramaster patagonicus and their anticancer activity. Mar. Drugs 2020, 18, 260. [Google Scholar] [CrossRef]
- Jin, W.; Rinehart, K.L.; Jares-Erijman, E.A. Ophidiacerebrosides: Cytotoxic glycosphingolipids containing a novel sphingosine from a sea star. J. Org. Chem. 1994, 59, 144–147. [Google Scholar] [CrossRef]
- Yamada, K.; Hara, E.; Miyamoto, T.; Higuchi, R.; Isobe, R.; Honda, S. Constituents of Holothuroidea, 6-Isolation and structure of biologically active glycosphingolipids from the sea cucumber Cucumaria echinata. Eur. J. Org. Chem. 1998, 371–378. [Google Scholar] [CrossRef]
- Higuchi, R.; Natori, T.; Komori, T. Biologically active glycosides from Asteroidea, XX. Glycosphingolipids from the starfish Asterina pectinifera, 1. Isolation and characterization of acanthacerebroside B and structure elucidation of related, nearly homogeneous cerebrosides. Liebigs Ann. Chem. 1990, 1, 51–55. [Google Scholar] [CrossRef]
- De Haan, J.W.; van de Ven, L.J.M. Configurations and Conformations in Acyclic, Unsaturated Hydrocarbons. A 13C NMR Study. Magn. Reson Chem. 1973, 5, 147–153. [Google Scholar] [CrossRef]
- Sugiyama, S.; Honda, M.; Komori, T. Biologically active glycosides from Asteroidea, XXIV 1). Stereochemistry of the four diastereomers of phytosphingosine. Liebigs Ann. Chem. 1990, 11, 1069–1078. [Google Scholar] [CrossRef]
- Horibata, S.; Vo, T.V.; Subramanian, V.; Thompson, P.R.; Coonrod, S.A. Utilization of the soft agar colony formation assay to identify inhibitors of tumorigenicity in breast cancer cells. J. Vis. Exp. 2015, 99, e52727. [Google Scholar] [CrossRef]
- AAT Bioquest. Available online: https://www.aatbio.com/tools/ic50-calculator (accessed on 10 November 2020).
- Kawatake, S.; Nakamura, K.; Inagaki, M.; Higuchi, R. Isolation and structure determination of six glucocerebrosides from the starfish Luidia maculata. Chem. Pharm. Bull. 2002, 50, 1091–1096. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH | δC | Position | δH | δC |
|---|---|---|---|---|---|
| 1a 1b | 4.51 dt (10.9, 4.2) 4.43 m | 61.8 | 11 | 2.10 q (6.7) | 27.3 |
| 1-OH | 6.60 m | 19 or 21 | 1.51 m | 27.9 | |
| 2 | 5.10 sext (4.5) | 52.8 | 20 or 22 | 0.88 d (6.5) | 22.5 |
| 3 | 4.34 m | 76.7 | 21 or 23 | 0.88 d (6.5) | 22.5 |
| 3-OH | 6.59 d (6.8) | NH | 8.55 d (9.0) | ||
| 4 | 4.28 m | 72.8 | 1′ | 175.0 | |
| 4-OH | 6.14 d (6.5) | 2′ | 4.62 m | 72.3 | |
| 5a 5b | 2.28 m 1.95 m | 33.9 | 2′-OH | 7.53 d (5.1) | |
| 6a 6b | 1.74 m 1.39 m | 26.1 | 3′a 3′b | 2.24 m 2.05 m | 35.5 |
| 7a 7b | 1.58 m 1.53 m | 30.2 | 4′a 4′b | 1.80 m 1.73 m | 25.6 |
| 8 | 2.17 q (7.0) | 27.5 | 14′ or 15′ | 1.25 m | 31.9 |
| 9 | 5.50 m | 130.1 | 15′ or 16′ | 1.27 m | 22.7 |
| 10 | 5.48 m | 129.9 | 16′ or 17′ | 0.89 t (7.2) | 14.0 |
| Position | 4 | 8, 9 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1a 1b | 4.70 dd (10.6, 5.6) 4.26 dd (10.6, 4.1) | 69.9 | 4.71 dd (10.6, 6.5) 4.53 dd (10.6, 4.4) | 70.3 |
| 2 | 4.81 m | 54.4 | 5.26 m | 51.5 |
| 3 | 4.77 m | 72.1 | 4.32 dd (12.3, 5.5) | 75.7 |
| 3-OH | 6.75 d (4.7) | 6.69 d (6.0) | ||
| 4 | 5.99 dd (15.8, 6.2) | 131.7 | 4.21 m | 72.4 |
| 5a 5b | 5.93 dt (15.8, 6.2) | 132.2 | 2.24 m 1.91 m | 33.9 |
| 6 | 2.12 m | 32.1 | ||
| 7 | 1.50 m | 29.4 | ||
| 8 | 2.11 m | 27.2 | ||
| 9 | 5.50 m | 130.3 | ||
| 10 | 5.49 m | 129.6 | ||
| 11 | 2.12 m | 27.0 | ||
| 19 or 14 | 1.30 m | 34.4 | 1.30 m | 34.5 |
| 20a or 15a 20b or 15b | 1.30 m 1.12 m | 36.7 | 1.30 m 1.10 m | 36.8 |
| 21 or 16 | 0.87 t (7.3) | 11.3 | 0.86 t (7.5) | 11.3 |
| 22 or 17 | 0.87 d (6.2) | 19.1 | 0.86 d (6.4) | 19.2 |
| NH | 8.32 d (9.0) | 8.54 d (9.2) | ||
| 1′ | 175.4 | 175.4 | ||
| 2′ | 4.58 m | 72.3 | 4.58 m | 72.3 |
| 2′-OH | 7.53 d (5.0) | 7.55 m | ||
| 3′a 3′b | 2.21 m 2.02 m | 35.4 | 2.20m 2.01 m | 35.4 |
| 4′a 4′b | 1.82 m 1.73 m | 25.7 | 1.77 m 1.70 m | 25.0 |
| Terminal CH3 | 0.89 t (7.0) | 14.0 | 0.88 t (7.0) | 14.0 |
| 1″ | 4.92 d (7.8) | 105.4 | 4.95 d (7.9) | 105.5 |
| 2″ | 4.03 m | 74.9 | 4.00 m | 74.9 |
| 2″-OH | 7.11 brs | |||
| 3″ | 4.20 m | 78.2 | 4.17 m | 78.2 |
| 4″ | 4.20 m | 71.4 | 4.17 m | 71.4 |
| 5″ | 3.91 m | 78.3 | 3.89 m | 78.3 |
| 6″a 6″b | 4.51 brd (12.5) 4.35 m | 62.5 | 4.48 brd (12.0) 4.33 m | 62.5 |
| 6″-OH | 6.26 brs | 6.27 brs | ||
| Compounds | Half Maximal Inhibitory Concentration (IC50), µM | |||
|---|---|---|---|---|
| HEK293 | HT-29 | SK-MEL-28 | MDA-MB-231 | |
| Doxorubicin | 35.7 ± 1.2 | 21.8 ± 3.2 | 40.0 ± 5.0 | 22.3 ± 0.2 |
| 1 | >50.0 >50.0 >50.0 >50.0 >50.0 >50.0 >50.0 | >50.0 >50.0 >50.0 >50.0 >50.0 >50.0 >50.0 | >50.0 >50.0 >50.0 >50.0 >50.0 >50.0 >50.0 | >50.0 |
| 2 | 48.7 ± 2.4 | |||
| 3 | 49.4 ± 1.6 | |||
| 5 | 48.7 ± 1.8 | |||
| 6 | 40.5 ± 0.5 | |||
| 7 | >50.0 | |||
| 9 | >50.0 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malyarenko, T.V.; Zakharenko, V.M.; Kicha, A.A.; Kuzmich, A.S.; Malyarenko, O.S.; Kalinovsky, A.I.; Popov, R.S.; Svetashev, V.I.; Ivanchina, N.V. New Ceramides and Cerebrosides from the Deep-Sea Far Eastern Starfish Ceramaster patagonicus. Mar. Drugs 2022, 20, 641. https://doi.org/10.3390/md20100641
Malyarenko TV, Zakharenko VM, Kicha AA, Kuzmich AS, Malyarenko OS, Kalinovsky AI, Popov RS, Svetashev VI, Ivanchina NV. New Ceramides and Cerebrosides from the Deep-Sea Far Eastern Starfish Ceramaster patagonicus. Marine Drugs. 2022; 20(10):641. https://doi.org/10.3390/md20100641
Chicago/Turabian StyleMalyarenko, Timofey V., Viktor M. Zakharenko, Alla A. Kicha, Alexandra S. Kuzmich, Olesya S. Malyarenko, Anatoly I. Kalinovsky, Roman S. Popov, Vasily I. Svetashev, and Natalia V. Ivanchina. 2022. "New Ceramides and Cerebrosides from the Deep-Sea Far Eastern Starfish Ceramaster patagonicus" Marine Drugs 20, no. 10: 641. https://doi.org/10.3390/md20100641
APA StyleMalyarenko, T. V., Zakharenko, V. M., Kicha, A. A., Kuzmich, A. S., Malyarenko, O. S., Kalinovsky, A. I., Popov, R. S., Svetashev, V. I., & Ivanchina, N. V. (2022). New Ceramides and Cerebrosides from the Deep-Sea Far Eastern Starfish Ceramaster patagonicus. Marine Drugs, 20(10), 641. https://doi.org/10.3390/md20100641