
Carbonic Anhydrase Inhibitors from Marine Natural Products


Abstract
1. Introduction
2. Sulfonamides
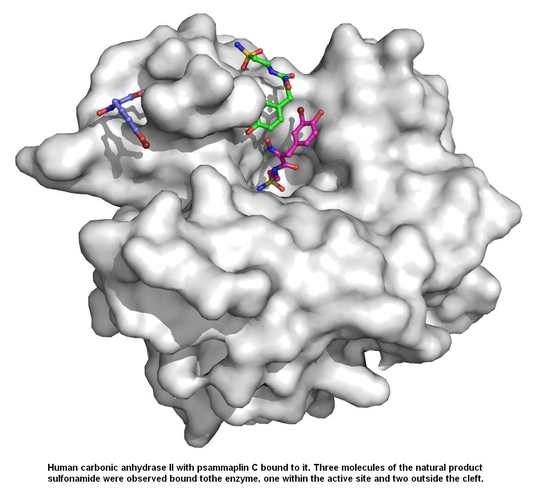
2.1. Psammaplin C
2.2. Altemicidin
3. Phenols
4. Polyamines
5. Coumarins
6. Miscellaneous Inhibitors
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ludwig, M. Carbonic anhydrase and the molecular evolution of C4 photosynthesis. Plant Cell Environ. 2012, 35, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Supuran, C.T. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: Can bacterial carbonic anhydrases shed new light on evolution of bacteria? J. Enzyme Inhib. Med. Chem. 2015, 30, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Aspatwar, A.; Tolvanen, M.E.E.; Barker, H.; Syrjänen, L.; Valanne, S.; Purmonen, S.; Waheed, A.; Sly, W.S.; Parkkila, S. Carbonic anhydrases in metazoan model organisms: Molecules, mechanisms, and physiology. Physiol. Rev. 2022, 102, 1327–1383. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023. [Google Scholar] [CrossRef]
- Supuran, C.T. Emerging role of carbonic anhydrase inhibitors. Clin. Sci. 2021, 135, 1233–1249. [Google Scholar] [CrossRef]
- Luca, V.D.; Vullo, D.; Scozzafava, A.; Carginale, V.; Rossi, M.; Supuran, C.T.; Capasso, C. An α-carbonic anhydrase from the thermophilic bacterium Sulphurihydrogenibium azorense is the fastest enzyme known for the CO2 hydration reaction. Bioorg. Med. Chem. 2013, 21, 1465–1469. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrases and Metabolism. Metabolites 2018, 8, 25. [Google Scholar]
- Supuran, C.T. Anti-obesity carbonic anhydrase inhibitors: Challenges and opportunities. J. Enzyme Inhib. Med. Chem. 2022, 37, 2478–2488. [Google Scholar]
- Giacomin, M.; Drummond, J.M.; Supuran, C.T.; Goss, G.G. The roles of plasma accessible and cytosolic carbonic anhydrases in bicarbonate (HCO3−) excretion in Pacific hagfish (Eptatretus stoutii). J. Comp. Physiol. B 2022, 192, 713–725. [Google Scholar]
- Rummer, J.L.; McKenzie, D.J.; Innocenti, A.; Supuran, C.T.; Brauner, C.J. Root effect hemoglobin may have evolved to enhance general tissue oxygen delivery. Science 2013, 340, 1327–1329. [Google Scholar] [CrossRef]
- McDonald, P.C.; Chafe, S.C.; Supuran, C.T.; Dedhar, S. Cancer Therapeutic Targeting of Hypoxia Induced Carbonic Anhydrase IX: From Bench to Bedside. Cancers 2022, 14, 3297. [Google Scholar] [CrossRef]
- Supuran, C.T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2017, 12, 61–88. [Google Scholar] [CrossRef]
- Nocentini, A.; Angeli, A.; Carta, F.; Winum, J.Y.; Zalubovskis, R.; Carradori, S.; Capasso, C.; Donald, W.A.; Supuran, C.T. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J. Enzyme Inhib. Med. Chem. 2021, 36, 561–580. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef]
- Liu, M.; Hansen, P.E.; Lin, X. Bromophenols in marine algae and their bioactivities. Mar. Drugs 2011, 9, 1273–1292. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Dong, S.; Erik Hansen, P.; Stagos, D.; Lin, X.; Liu, M. Progress of Bromophenols in Marine Algae from 2011 to 2020: Structure, Bioactivities, and Applications. Mar. Drugs 2020, 18, 411. [Google Scholar] [CrossRef]
- Mujumdar, P.; Poulsen, S.A. Natural Product Primary Sulfonamides and Primary Sulfamates. J. Nat. Prod. 2015, 78, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Schinke, C.; Martins, T.; Queiroz, S.C.N.; Melo, I.S.; Reyes, F.G.R. Antibacterial Compounds from Marine Bacteria, 2010–2015. J. Nat Prod. 2017, 80, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase activators. Future Med. Chem. 2018, 10, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Novel carbonic anhydrase inhibitors. Future Med. Chem. 2021, 13, 1935–1937. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, D.P.; Seleem, M.N.; Supuran, C.T. Bacterial carbonic anhydrases: Underexploited antibacterial therapeutic targets. Future Med. Chem. 2021, 13, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, C.; Crews, P. Novel marine sponge derived amino acids 13. Additional psammaplin derivatives from Psammaplysilla purpurea. Tetrahedron 1991, 47, 2097–2102. [Google Scholar] [CrossRef]
- Mujumdar, P.; Teruya, K.; Tonissen, K.F.; Vullo, D.; Supuran, C.T.; Peat, T.S.; Poulsen, S.A. An Unusual Natural Product Primary Sulfonamide: Synthesis, Carbonic Anhydrase Inhibition, and Protein X-ray Structures of Psammaplin C. J. Med. Chem. 2016, 59, 5462–5470. [Google Scholar] [CrossRef]
- Mujumdar, P.; Kopecka, J.; Bua, S.; Supuran, C.T.; Riganti, C.; Poulsen, S.A. Carbonic Anhydrase XII Inhibitors Overcome Temozolomide Resistance in Glioblastoma. J. Med. Chem. 2019, 62, 4174–4192. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors: An update on experimental agents for the treatment and imaging of hypoxic tumors. Expert Opin. Investig. Drugs 2021, 30, 1197–1208. [Google Scholar] [CrossRef]
- Supuran, C.T. Experimental Carbonic Anhydrase Inhibitors for the Treatment of Hypoxic Tumors. J. Exp. Pharmacol. 2020, 12, 603–617. [Google Scholar] [CrossRef]
- Takahashi, A.; Kurasawa, S.; Ikeda, D.; Okami, Y.; Takeuchi, T. Altemicidin, a new acaricidal and antitumor substance. I. Taxonomy, fermentation, isolation and physico-chemical and biological properties. J. Antibiot. 1989, 42, 1556–1561. [Google Scholar] [CrossRef]
- Simonsson, I.; Jonsson, B.H.; Lindskog, S. Phenol, a competitive inhibitor of CO2 hydration catalyzed by carbonic anhydrase. Biochem. Biophys. Res. Commun. 1982, 108, 1406–1412. [Google Scholar] [CrossRef]
- Nair, S.K.; Ludwig, P.A.; Christianson, D.W. Two-site binding of phenol in the active site of human carbonic anhydrase II: Structural implications for substrate association. J. Am. Chem. Soc. 1994, 116, 3659–3660. [Google Scholar] [CrossRef]
- Innocenti, A.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Interactions of phenols with the 12 catalytically active mammalian isoforms (CA I–XIV). Bioorg. Med. Chem. Lett. 2008, 18, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, A.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I–XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg. Med. Chem. 2008, 16, 7424–7428. [Google Scholar] [CrossRef] [PubMed]
- Bayram, E.; Senturk, M.; Kufrevioglu, O.I.; Supuran, C.T. In vitro effects of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymesI and II. Bioorg. Med. Chem. 2008, 16, 9101–9105. [Google Scholar] [CrossRef] [PubMed]
- Şentürk, M.; Gülçin, I.; Daştan, A.; Küfrevioğlu, Ö.I.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg. Med. Chem. 2009, 17, 3207–3211. [Google Scholar] [CrossRef]
- Innocenti, A.; Öztürk Sarıkaya, S.B.; Gülçin, I.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I–XIV with a series of natural product polyphenols and phenolic acids. Bioorg. Med. Chem. 2010, 18, 2159–2164. [Google Scholar] [CrossRef]
- Davis, R.A.; Innocenti, A.; Poulsen, S.A.; Supuran, C.T. Carbonic anhydrase inhibitors. Identification of selective inhibitors of the human mitochondrial isozymes VA and VB over the cytosolic isozymes I and II from a natural product-based phenolic library. Bioorg. Med. Chem. 2010, 18, 14–18. [Google Scholar] [CrossRef]
- Davis, R.A.; Hofmann, A.; Osman, A.; Hall, R.A.; Mühlschlegel, F.A.; Vullo, D.; Innocenti, A.; Supuran, C.T.; Poulsen, S.A. Natural product-based phenols as novel probes for mycobacterial and fungal carbonic anhydrases. J. Med. Chem. 2011, 54, 1682–1692. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibition with natural products: Novel chemotypes and inhibition mechanisms. Mol. Divers 2011, 15, 305–316. [Google Scholar] [CrossRef]
- Karioti, A.; Ceruso, M.; Carta, F.; Bilia, A.R.; Supuran, C.T. New natural product carbonic anhydrase inhibitors incorporating phenol moieties. Bioorg. Med. Chem. 2015, 23, 7219–7225. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. The diversity of naturally occurring organobromine compounds. Chem. Soc. Rev. 1999, 28, 335–346. [Google Scholar] [CrossRef]
- Balaydın, H.T.; Sentürk, M.; Menzek, A. Synthesis and carbonic anhydrase inhibitory properties of novel cyclohexanonyl bromophenol derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Balaydin, H.T.; Durdaği, S.; Ekinci, D.; Sentürk, M.; Göksu, S.; Menzek, A. Inhibition of human carbonic anhydrase isozymes I, II and VI with a series of bisphenol, methoxy and bromophenol compounds. J. Enzyme Inhib. Med. Chem. 2012, 27, 467–475. [Google Scholar] [CrossRef]
- Balaydın, H.T.; Şentürk, M.; Göksu, S.; Menzek, A. Synthesis and carbonic anhydrase inhibitory properties of novel bromophenols and their derivatives including natural products: Vidalol B. Eur. J. Med. Chem. 2012, 54, 423–428. [Google Scholar] [CrossRef]
- Taslimi, P.; Gülçin, İ.; Öztaşkın, N.; Çetinkaya, Y.; Göksu, S.; Alwasel, S.H.; Supuran, C.T. The effects of some bromophenols on human carbonic anhydrase isoenzymes. J. Enzyme Inhib. Med. Chem. 2016, 31, 603–607. [Google Scholar] [CrossRef]
- Boztaş, M.; Çetinkaya, Y.; Topal, M.; Gülçin, İ.; Menzek, A.; Şahin, E.; Tanc, M.; Supuran, C.T. Synthesis and carbonic anhydrase isoenzymes I, II, IX, and XII inhibitory effects of dimethoxybromophenol derivatives incorporating cyclopropane moieties. J. Med. Chem. 2015, 58, 640–650. [Google Scholar] [CrossRef]
- Carta, F.; Temperini, C.; Innocenti, A.; Scozzafava, A.; Kaila, K.; Supuran, C.T. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J. Med. Chem. 2010, 53, 5511–5522. [Google Scholar] [CrossRef]
- Davis, R.A.; Vullo, D.; Supuran, C.T.; Poulsen, S.A. Natural product polyamines that inhibit human carbonic anhydrases. Biomed. Res. Int. 2014, 2014, 374079. [Google Scholar] [CrossRef]
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef]
- Supuran, C.T. Coumarin carbonic anhydrase inhibitors from natural sources. J. Enzyme Inhib. Med. Chem. 2020, 35, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-zinc mediated inhibition of carbonic anhydrases: Coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J. Med. Chem. 2010, 53, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Vullo, D.; Maresca, A.; Supuran, C.T.; Poulsen, S.A. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, K.; Khan, A.; Ur Rehman, N.; Halim, S.A.; Khan, M.; Ali, L.; Hilal Al-Balushi, A.; Al-Busaidi, H.K.; Al-Harrasi, A. New Carbonic Anhydrase-II Inhibitors from Marine Macro Brown Alga Dictyopteris hoytii Supported by In Silico Studies. Molecules 2021, 26, 7074. [Google Scholar] [CrossRef] [PubMed]
- Artasensi, A.; Angeli, A.; Lammi, C.; Bollati, C.; Gervasoni, S.; Baron, G.; Matucci, R.; Supuran, C.T.; Vistoli, G.; Fumagalli, L. Discovery of a Potent and Highly Selective Dipeptidyl Peptidase IV and Carbonic Anhydrase Inhibitor as “Antidiabesity” Agents Based on Repurposing and Morphing of WB-4101. J. Med. Chem. 2022, 65, 13946–13966. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| hCA Isoform | KI (nM) | |
| 1 | AZA | |
| hCA I | 48.1 | 250 |
| hCA II | 88.0 | 12 |
| hCA IV | 75.3 | 74.2 |
| hCA VA | 154 | 63.1 |
| hCA VI | 9680 | 11.0 |
| hCA VII | 1.7 | 2.5 |
| hCA IX | 12.3 | 25.0 |
| hCA XII | 0.79 | 5.7 |
| hCA XIII | 10630 | 16.4 |
| hCA XIV | 379 | 41.3 |
 | ||||||
|---|---|---|---|---|---|---|
| Polyamine | KI (μM) | |||||
| hCA I | hCA II | hCA IV | hCA IX | hCA XII | hCA XIV | |
| Spermine | 231 | 84 | 0.010 | 13.3 | 27.6 | 0.86 |
| Spermidine | 1.40 | 1.11 | 0.112 | 1.37 | 44.1 | 1.00 |
| 28 | 1.76 | 0.41 | 6.72 | 0.20 | 2.81 | 2.12 |
| 29 | 0.77 | 0.37 | 9.10 | 0.35 | 3.48 | 2.28 |
| 30 | 0.86 | 0.35 | 9.08 | 0.27 | 3.50 | 6.96 |
| 31 | 0.85 | 0.48 | >20 | 0.34 | >20 | 2.72 |
| 32 | 0.79 | 0.34 | 7.03 | 0.36 | 4.21 | 1.52 |
| AAZ | 0.25 | 0.012 | 0.074 | 0.025 | 0.006 | 0.041 |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | KI (μM) | |||||
| hCA I | hCA II | hCA VII | hCA IX | hCA XII | hCA XIII | |
| 37 | 6.45 | >100 | 14.5 | 3.22 | 9.07 | 4.63 |
| 38 | 6.55 | >100 | 78.4 | 3.27 | 1.79 | 4.24 |
| 39 | 40.1 | >100 | 58.3 | 6.33 | 8.51 | 3.70 |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | KI (μM) | |||||
| Rv3273 | Rv1284 | Nce103 | Can2 | hCA I | hCA II | |
| 40 | 0.91 | 11.8 | 0.92 | 0.89 | 10.5 | 9.6 |
| 41 | 0.92 | 0.91 | 0.90 | 0.95 | 355 | 13.1 |
| Phenol | 79.0 | 64.0 | 17.3 | 25.9 | 10.1 | 5.5 |
| AZA | 0.10 | 0.48 | 0.13 | 0.01 | 0.25 | 0.012 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Supuran, C.T. Carbonic Anhydrase Inhibitors from Marine Natural Products. Mar. Drugs 2022, 20, 721. https://doi.org/10.3390/md20110721
Supuran CT. Carbonic Anhydrase Inhibitors from Marine Natural Products. Marine Drugs. 2022; 20(11):721. https://doi.org/10.3390/md20110721
Chicago/Turabian StyleSupuran, Claudiu T. 2022. "Carbonic Anhydrase Inhibitors from Marine Natural Products" Marine Drugs 20, no. 11: 721. https://doi.org/10.3390/md20110721
APA StyleSupuran, C. T. (2022). Carbonic Anhydrase Inhibitors from Marine Natural Products. Marine Drugs, 20(11), 721. https://doi.org/10.3390/md20110721