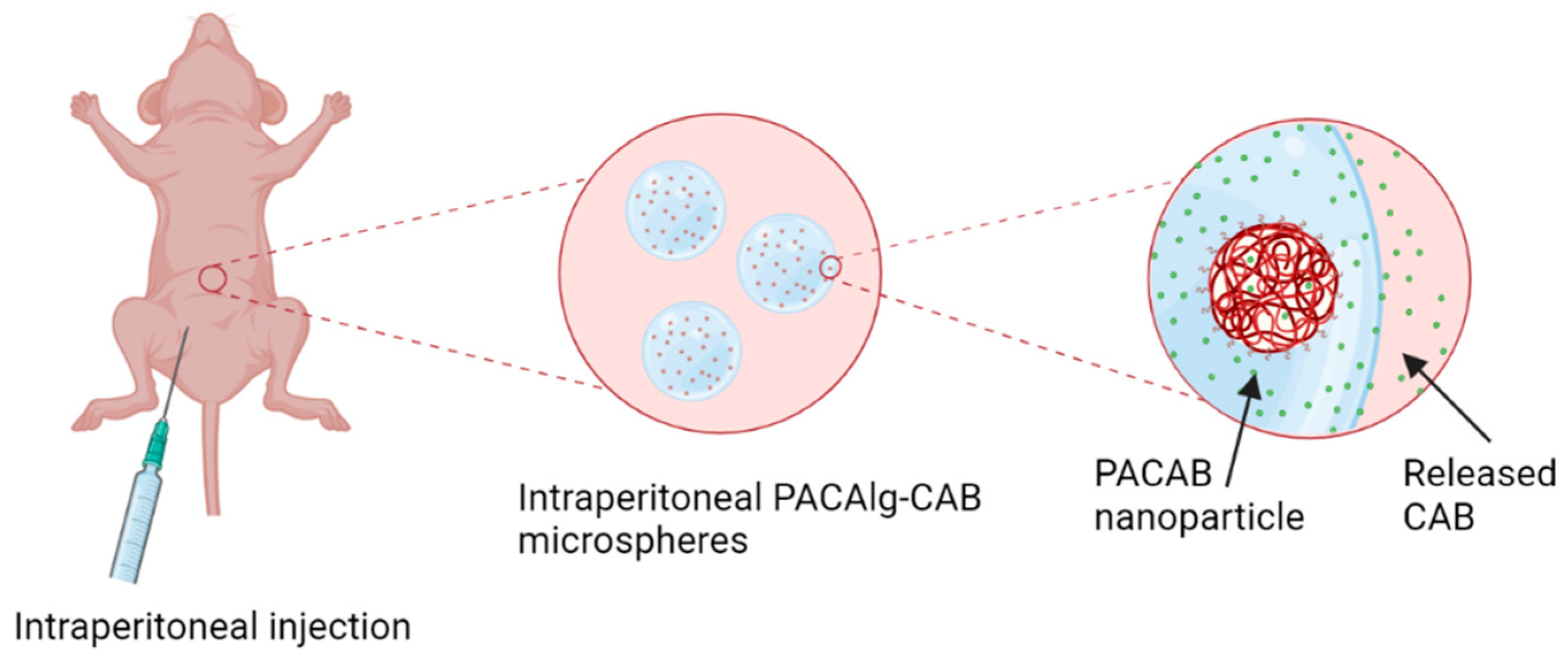
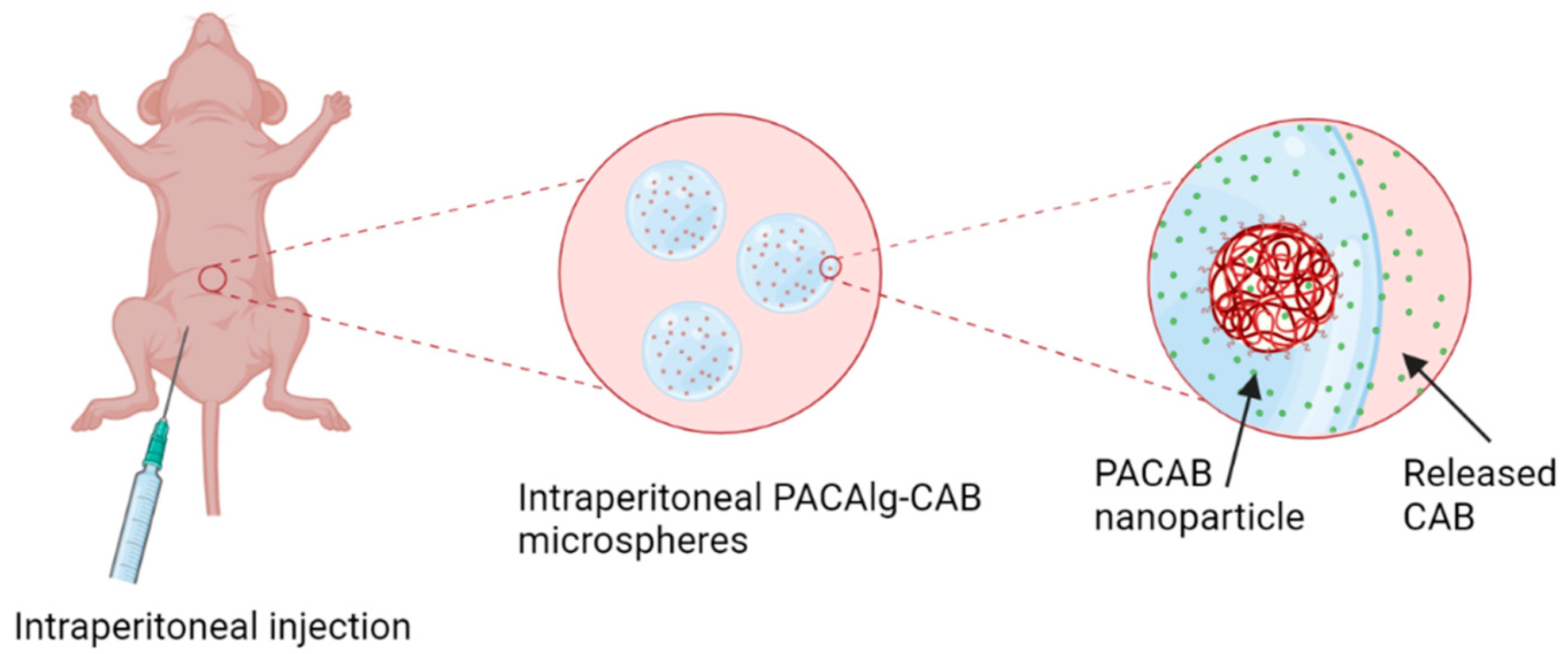
1. Introduction
Microparticulate drug delivery systems offer numerous advantages in drug administration due to their structural and functional properties, including improved drug stability, controlled and sustained drug release, reduced drug toxicity, and target specificity [
1]. Microparticles can be administrated by several routes (e.g., by oral ingestion, inhalation, topical use, or direct injection into the blood or specific tissues) and have been shown to have a strong therapeutic impact for many indications, including cancer, diabetes, cardiovascular diseases and neurological disorders [
1,
2]. The particle material is usually based on biocompatible polymers, where the alginate polysaccharide family is shown to be one of the biopolymers with the widest biomedical applicability [
3]. Alginates are naturally occurring linear polymers derived from the cell walls of brown algae, that crosslink with divalent cations to form highly porous hydrogels with an inert aqueous environment within the gel matrix [
4]. Due to their low toxicity and immunogenicity profile, alginate hydrogels have been broadly investigated for use in biomedical applications, including encapsulation of islet cells for diabetes type 1 treatment, wound healing, and cancer treatment [
3,
5,
6,
7].
The peritoneal cavity is a common metastatic site for malignancies of abdominopelvic organs, and is associated with a poor prognosis [
8,
9]. Cytoreductive surgery (CRS) followed by hyperthermic intraperitoneal chemotherapy (HIPEC) is the only curative treatment currently available for these patients. This treatment is, however, only suitable for a subgroup of patients and many of them experience a relapse—highlighting the need for new treatment options [
10,
11]. These patients respond poorly to systemic therapy, and intraperitoneal administration of cytotoxic drugs has therefore been used to achieve high local drug concentrations. The drugs are, however, rapidly cleared from the peritoneal cavity [
12]. Hence, there is a need for therapeutic approaches that prolong the residence time of the drugs in the peritoneal cavity to increase drug exposure to peritoneal tumors [
13]. This can be achieved by encapsulation of drugs into carriers, to both increase drug retention and reduce systemic toxicity. We have previously shown promising drug retention and improved treatment efficacy by encapsulating cabazitaxel (CAB) in poly(alkyl cyanoacrylate) (PACA) nanoparticles (NPs) [
14]. CAB is a second-generation taxane approved for the treatment of patients with hormone-refractory metastatic prostate cancer [
15]. This PACAB platform was shown to be a robust drug delivery system for treatment of peritoneal cancers, and is currently under commercial development. CAB has previously been nanoformulated in liposomes, lipospheres and PACA NPs for cancer treatment [
16,
17,
18,
19,
20], but this was, to our knowledge, the first work presenting CAB-loaded NPs for treatment of intraperitoneal cancers. To potentially prolong drug retention in the peritoneal cavity, we hypothesized that encapsulation of the PACAB NPs into alginate microspheres would improve the balance between therapeutic efficacy and toxicity.
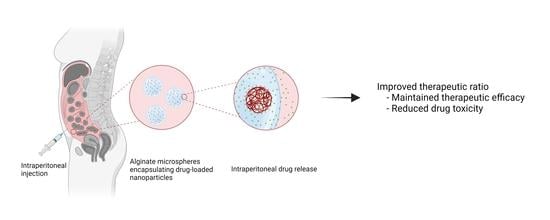
In this study, we present the PACAlg technology, where alginate microparticles encapsulating PACAB NPs were designed and synthesized to improve intraperitoneal drug retention and reduce drug toxicity (
Figure 1). The technology was evaluated by physicochemical characterization of the PACAlg microparticles, in vitro cytotoxicity studies, and finally, CAB biodistribution and treatment efficacy studies were conducted in patient-derived xenograft (PDX) models mimicking peritoneal metastasis.
3. Discussion
Peritoneal metastases are associated with poor prognosis, and more efficacious treatment options are needed [
10,
11]. Drug encapsulation in NPs has previously shown promising effects by delaying systemic drug absorption through prolongation of the peritoneal residence time [
13,
21,
22]. Due to their physicochemical characteristics, NPs are less prone to clearance from the peritoneal cavity, and the increased retention leads to higher local drug concentrations that can result in improved treatment efficacy. In this study, novel biocompatible alginate microspheres encapsulating drug-loaded NPs (PACAlg-CAB) were developed with the aim to further increase peritoneal retention and reduce toxic side effects, and hence improving cancer treatment efficacy of cytostatic drugs. To our knowledge, this is the first time alginate has been used as a microparticulate carrier to encapsulate NPs for the treatment of intraperitoneal cancer.
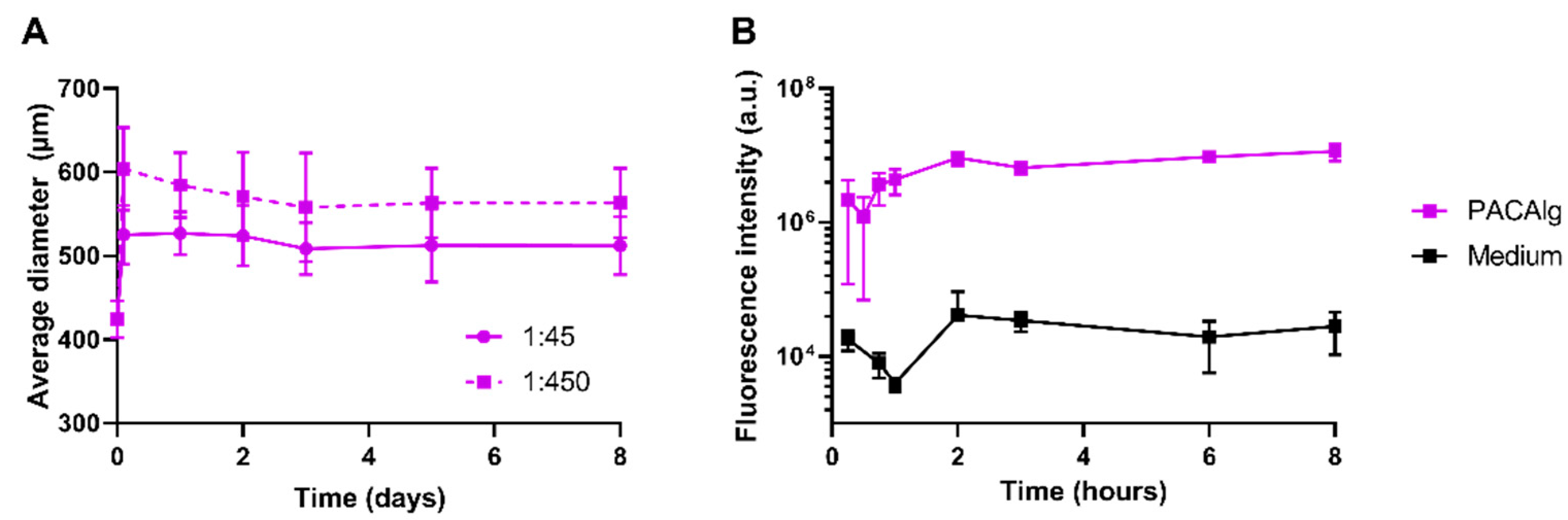
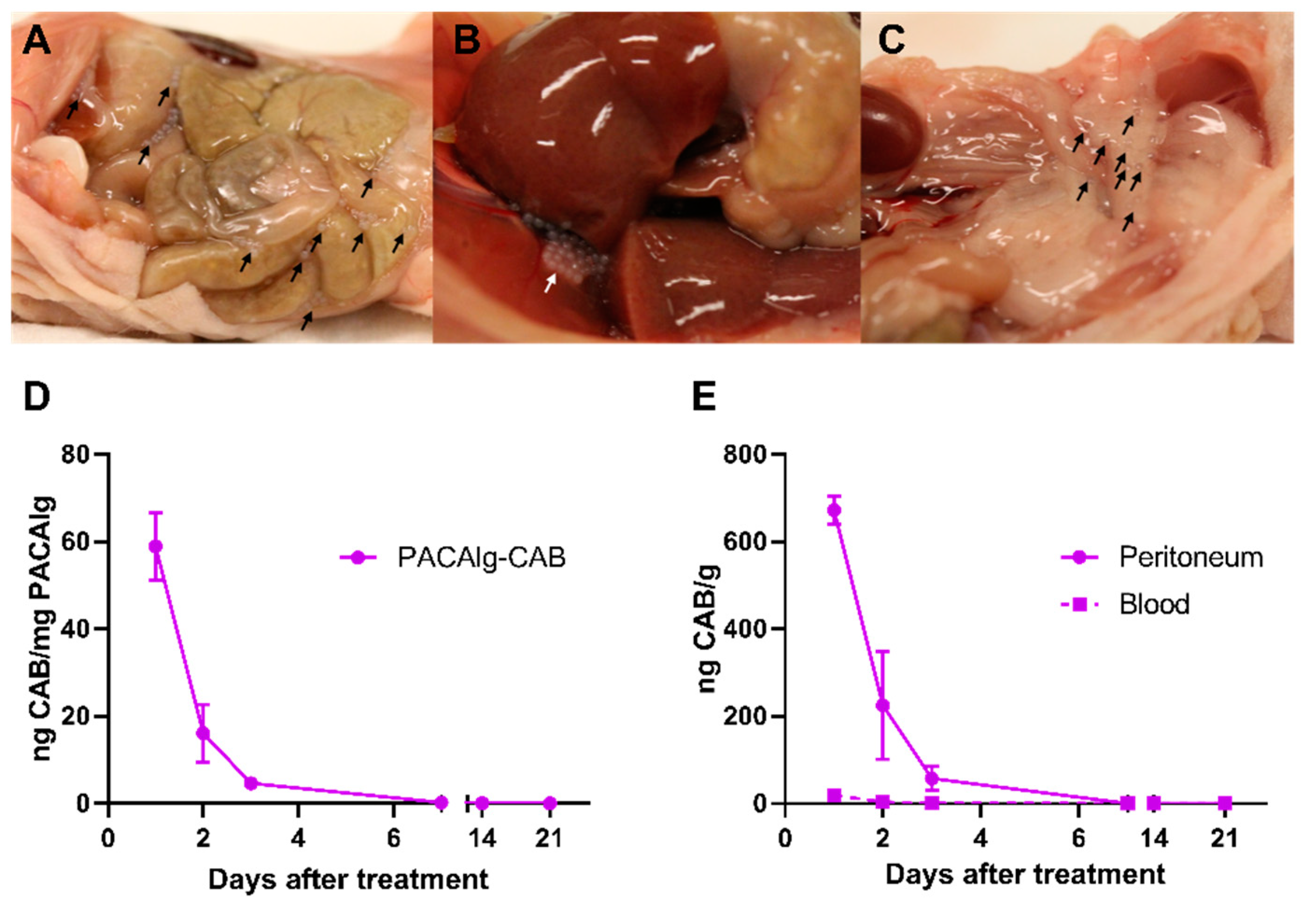
Electrostatic droplet generation was used to synthesize spherical and monodisperse PACAlg alginate hydrogels with an average diameter of approximately 450 µm. The microspheres were shown to be stable with respect to size and morphology for at least 8 days in physiological conditions in vitro. Fluorescence stability over the same period indicated that the alginate forms rigid and stable crosslinked networks that retain the fluorescently labeled NPs within the microspheres, even after initial osmotic swelling. This shows that NP degradation is needed in order to release the active pharmaceutical ingredient (API) from the microspheres. The in vivo biodistribution study gave similar results, showing microsphere stability for at least 21 days after injection in the peritoneal cavity in mice. This demonstrates that alginate forms crosslinked hydrogel networks that are stable also at in vivo conditions.
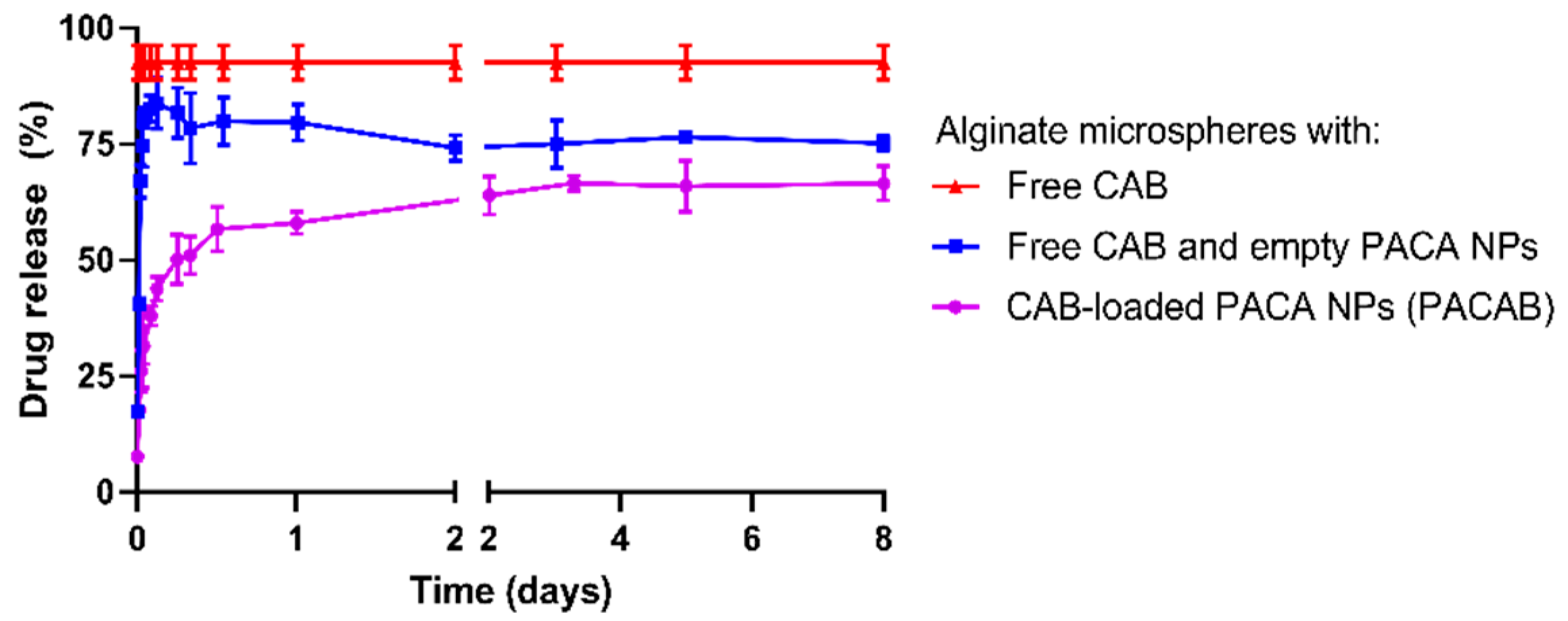
Drug release data showed that the alginate microspheres alone have limited retention capacities on the CAB drug. CAB is a relatively small hydrophobic drug (836 Da), which is not expected to interact with the highly hydrophilic alginate backbone or be retained in the alginate porous network [
23]. Co-encapsulation with empty NPs showed a slight restriction of CAB release, suggesting an increased interaction between CAB and the NP/hydrogel matrix compared to that of CAB and the pure alginate gel. This was not expected, as the hydrophilic surfaces of PEGylated NPs were thought to have a minimal affinity towards CAB. The observed retention could therefore be a consequence of NP degradation, with CAB showing affinity to hydrophobic components of the PACA polymers or their degradation products. This may also explain the lack of complete (100%) drug release seen in vitro for both the PACAlg-CAB (67%) and PACAlg with co-encapsulated CAB (75%) samples.
PACAlg-CAB drug release data showed a gradual drug release over the first two days in vitro, before the curve reached the plateau. The release rate could possibly be further prolonged by using NPs with an inherently longer drug release rate, such as a PACA polymer with longer alkyl side-chains [
24]. PACAlg drug release could also be tuned by editing the chemical composition of the alginate hydrogel (the secondary barrier) by introducing polymers or polymer side chains with a varying affinity towards the API. It was previously shown that cyclodextrin-grafted alginates can modulate the drug release of paclitaxel [
25]—a secondary barrier that potentially also could be applied to the PACAlg drug delivery system.
As shown by the fluorescence stability study, NPs were not released from the microspheres within the observation period. One can thereby assume drug release as a free drug only—firstly as a release from the PACA NPs, secondly as diffusion out of the alginate network. This eliminates cellular uptake of NP-encapsulated drugs, and leaves diffusion of the hydrophobic drug across the cellular membranes as the most probable drug delivery mechanism.
Even though NP-based drug encapsulation for peritoneal administration is an appealing strategy, it may involve toxic effects, including NPs entering the systemic circulation, accumulation of NPs in the spleen and liver, and inflammatory responses in the peritoneum [
26]. NPs of various sizes and materials have also been shown to cause ascites production in tumor-bearing mice, resulting in reduced survival [
27]. New strategies to reduce the toxic effects are therefore needed, and it was hypothesized that NP encapsulation in alginate microspheres could reduce the toxic potential of intraperitoneal NPs. Alginate has commonly been explored for intraperitoneal administration, e.g., for encapsulation of insulin-producing islet cells and hepatocytes [
28,
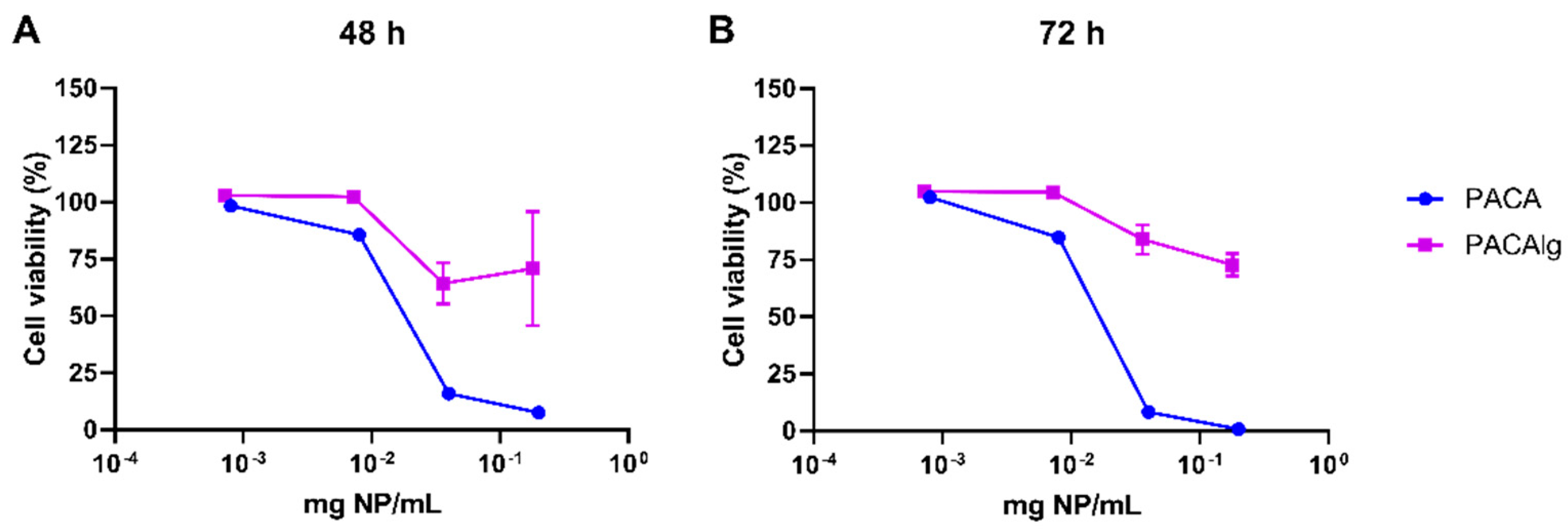
29]. The in vitro cytotoxicity results presented in the current study show promising effects of alginate encapsulation. The alginate hydrogel was non-toxic for normal mesothelial cells (LP-9), and encapsulation reduced the toxic effects of the NP material. Mesothelial cells line the surface of the peritoneal cavity and are therefore a highly relevant cell model for toxicity evaluation of novel intraperitoneal treatment strategies.
In this study, the taxane CAB was used as the API for cancer therapy. CAB is a highly potent chemotherapeutic agent with the drawback of inducing severe toxic side effects in healthy tissues [
15]. Encapsulation of CAB into nanocarriers is shown to reduce these side effects, and also prolong blood circulation half-lives [
30]—allowing for efficient preclinical antitumor effects after dose regimens of 6–15 mg/kg in mice [
18,
19]. For APIs where high cumulative doses are required for treatment effect, e.g., doxorubicin [
31], a higher number of NPs would be needed to deliver the necessary doses. The observed reduction of NP-based toxic side effects by alginate encapsulation could therefore be essential to allow the administration of adequate doses of other APIs, thereby allowing novel treatment strategies for these drugs.
Peritoneal metastases may be located throughout the entire peritoneal cavity, representing a large surface area, as the peritoneal area is equivalent to the body surface area in humans [
32,
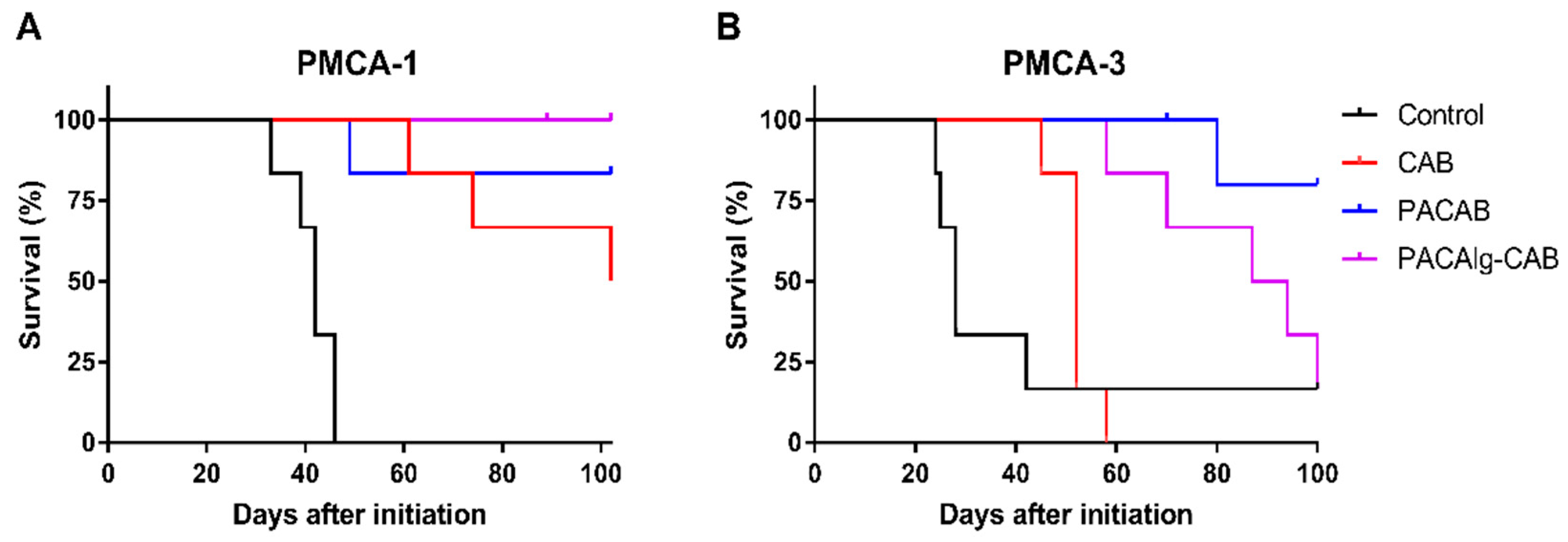
33]. To achieve optimal treatment efficacy, it is therefore important that the cytotoxic drugs are distributed throughout the peritoneal cavity to ensure drug exposure to all metastases. PACAlg-CAB microspheres were shown to be well distributed throughout the peritoneal cavity, without adhesion to tissue surfaces. The therapeutic potential of PACAlg-CAB was evaluated in two relevant PDX models mimicking peritoneal metastasis. The PMCA-1 model was highly CAB sensitive, and no significant improvement in survival was observed by any of the NP-based delivery systems. Interestingly, however, all PACAlg-CAB-treated mice were cured by the treatment, while for the other groups some mice eventually had to be sacrificed because of peritoneal metastases. In the PMCA-3 model, both PACAB and PACAlg-CAB significantly improved survival compared to CAB alone. This is a high-grade tumor with signet cell differentiation, which is known for being an aggressive phenotype that is hard to treat. The in vivo results clearly show that the treatment efficacy of CAB was at least maintained by NP encapsulation. However, incorporating PACAB into the alginate microspheres did not further increase drug efficacy, and for CAB, in particular, the additional retention and reduced toxicity may not be necessary. However, for drugs with high unspecific toxicity that must be delivered at high doses, for drugs where the NP loading capacity is lower than for CAB, where high amounts of NP must be delivered, the PACAlg drug delivery platform could still represent an important improvement.
In conclusion, this proof-of-concept study presents the alginate encapsulation of drug-loaded NPs as a promising novel strategy for drug administration in the peritoneal cavity that could improve the therapeutic ratio of toxic drugs. PACAlg allows for reduced cytotoxicity, the possibility of administering higher drug doses, and an increased ability to tune drug release rates compared to that of NPs alone. The technology could be further improved by exchanging the API or by tuning the drug release rate by chemical modification of the alginate hydrogel.
4. Materials and Methods
4.1. Synthesis and Characterization of PACA Nanoparticles
PACA NPs were synthesized using the miniemulsion polymerization technique, as previously described [
19,
34,
35]. Briefly, oil-in water-emulsions were prepared by mixing a monomer oil phase with an acidic water phase. The oil phase contained ethylbutyl-cyanoacrylate (EBCA; Cuantum Medical Cosmetics, Bellaterra, Spain), the co-stabilizer MIGLYOL
® 812 N (2.3% (
w/
w), Cremer OLEO GmbH & Co. KG, Hamburg, Germany), and the payload (0.2% (
w/
w) of the fluorescent dye NR668 (modified Nile Red, custom synthesis [
36])) and/or the cytostatic agent CAB (10.2% (
w/
w); Biochempartner Ltd., Wuhan, China)). The water phase consisted of the non-ionic PEG stabilizers Brij
® L23 (6.8 mM, Sigma-Aldrich, St. Louis, MO, USA) and Kolliphor
® HS 15 (8.7 mM in 0.1 M HCl; Sigma-Aldrich (St. Louis, MO, USA)). The emulsions were sonicated (50% amplitude, 450 Digital Sonifier
®, Branson Ultrasonics, Danbury, CT, USA) in an ice bath for 3 min, and subsequently stirred overnight at room temperature. The pH was adjusted to 5 with 0.1 M NaOH, and the stirring was continued for at least 5 h. Dialysis (Spectra/Por dialysis membrane, 12–14 kDa MWCO; Spectrum Labs, Rancho Dominguez, CA, USA) against 1 mM HCl was performed to remove any excess stabilizers from the NP solutions.
The synthesized NPs were characterized for size (Z-average), polydispersity index (PDI), and surface charge (zeta potential) using electrophoretic and dynamic light scattering (ELS and DLS) (Zetasizer Nano ZS, Malvern Instruments/Malvern Panalytical, Malvern, UK) in 0.01 M phosphate buffer, pH 7.
4.2. Synthesis and Characterization of PACAlg Microspheres
PACAlg microspheres were produced by an extrusion-dripping technique using an electrostatic bead generator (the prototype designed by the Trondheim Bioencapsulation Group, NTNU, and the commercially available version from Nisco, Zurich, Switzerland) [
37]. First, a sodium alginate solution (2.2% (
w/
v) PRONOVA™ UP LVG, pH 5; NovaMatrix, Sandvika, Norway) was mixed with the PACA NP solution to form a suspension with alginate concentration of 1.8% (
w/
v) and 18% (
v/
v) NP stock solution. The suspension was stirred overnight at 4 °C to form a homogeneous suspension and to release air bubbles. Microspheres were synthesized by extruding the alginate solution through a nozzle (350 µm outer diameter, 170 µm inner diameter) by using a syringe pump (Cole-Parmer, Vernon Hills, IL, USA) at a flow rate of 10 mL/h, an applied voltage of 7.0 kV and a 4.5 cm collecting distance down to a calcium chloride (50 mM, pH 5; Sigma-Aldrich (St. Louis, MO, USA), hardening bath where the microspheres were left to crosslink for 10 min at room temperature under magnetic stirring. Empty microspheres, microspheres encapsulating free drug, and microspheres co-encapsulating free drug together with empty NPs were synthesized as controls.
The size and shape of the microspheres were characterized by phase contrast and fluorescence microscopy (Nikon Eclipse TS100 with a Plan Flour 4X/0.13 NA objective with a pE-4000 CoolLED illumination system and a TRITC Quad filter set, λ
ex = 550 nm; Nikon, Tokyo, Japan). ImageJ2 [
38] was used to determine the average diameter of 40 microspheres imaged by phase contrast.
Aspect ratio (AR) and
sphericity factor (SF) were calculated for the same microspheres by using the Analyze particles function in ImageJ2 and the following equations:
where
DMax is the maximum Feret diameter,
DOrthogonal is the orthogonal diameter and
DMin is the minimum Feret diameter [
39]. Microspheres with
AR < 1.1 and
SF < 0.05 were considered spherical. Swelling behavior and size stability were studied by incubating microspheres in PBS with calcium chloride and magnesium chloride (1:450 dilution factor; D1283, Sigma-Aldrich (St. Louis, MO, USA), pH 7.4 at 37 °C in the dark.
The NP distribution in the alginate microspheres was examined by confocal laser scanning microscopy (CLSM), using a Leica SP8 microscope with a 25X/0.95 NA water objective and a 20X/0.75 NA dry immersion objective (Leica Microsystems, Wetzlar, Germany). The NP-encapsulated fluorophore NR668 was excited by a white light laser at 534 nm, and emission was detected at 574–627 nm.
The retention of NPs within the alginate microspheres was quantified by measuring the fluorescence intensity of microspheres filled with dye-loaded NPs. The microspheres were diluted (1:450 volumetric dilution factor) in PBS with calcium chloride and magnesium chloride, pH 7.4, at 37 °C in the dark, and analyzed on a microplate reader (SpectraMax i3x, Molecular Devices, San Jose, CA, USA, λ
ex = 514 nm, λ
em = 600 nm). The retention of NPs was calculated as follows:
where
FInitial is the initial fluorescence intensity of the incubated microspheres and
FMeasured is the fluorescence intensity of the medium at specific time points.
Drug release from the microspheres was examined by incubating microspheres loaded with PACAB in PBS with calcium chloride and magnesium chloride (1:450 dilution factor), pH 7.4 and supplemented with 0.5% (
w/
v) Tween 80 to ensure drug solubility. Aliquots (100 µL) were withdrawn at predetermined time points, diluted 1:100 in acetone, and analyzed for drug content by liquid chromatography tandem mass spectrometry (LC-MS/MS) as described below. The experiments were performed in independent triplicates, and conducted at 37 °C in the dark under mild stirring for up to 8 days. Two control groups: I: microspheres encapsulating free drug and II: free drug mixed with microspheres encapsulating empty NPs were included in the experiment. Percentage drug release was determined using the following equation:
where
CMax is estimated by:
where
CAlg and
VAlg are the calculated drug concentration and volume of the alginate solution used for encapsulation, respectively.
CGel and
VGel are the quantified drug concentration and volume of the alginate gelling solution, respectively.
VMedium denotes the volume of the PBS incubation medium. The release data was fitted to an exponential release model with a nonlinear least square approach, using the Curve Fitting Toolbox in Matlab (Release 2018a, The MatWorks, Inc., Natick, MA, USA). Data was fitted to the following equation:
where Release (
t) is the percentage release at time
t after microsphere incubation,
K0 the instantaneous release at
t = 0, k the release rate constant and
K +
K0 the steady-state conditions.
4.3. CAB Quantification by LC-MS/MS
CAB content was quantified by LC-MS/MS as previously described [
18], using an Agilent 1290 HPLC system coupled to an Agilent 6495 triple quadrupole mass spectrometer, equipped with an Agilent Jet Stream ion source (Agilent Technologies, Santa Clara, CA, USA). CAB was chromatographically separated from the matrix using an Ascentis
® Express C8 column (75 × 2.1 mm, 2.7 μm particle size), with a 5 × 2.1 mm guard column of the same material (Sigma-Aldrich, St. Louis, MO, USA), at 40 °C. Mobile phase A was 25 mM formic acid in water and Mobile phase B was 100% methanol, run at a flow rate of 0.5 mL/min. The mobile phase gradient was isocratic at 55% B for 1.5 min, then increased to 80% B over 1 minute, followed by 1 min washout time and subsequently column re-equilibration. This resulted in a retention time of 3.1 min and a run time of 5.0 min per injection. Injection volume was 5 μL. MS detection was in positive mode, quantified in multiple reaction monitoring (MRM) mode using the transition
m/z 858.3 → 577.2. The parent ion was chosen to be the Na adduct as this gave the best sensitivity. Similarly, the hexadeuterated internal standard was detected on the 864.4 → 583.2 transition. Both analytes were run at 380 V fragmentor and 20 V collision energy voltages.
Reference standards were used to ensure accurate quantification. The unlabelled CAB standard used for NP encapsulation was dissolved in acetone to 1 mg/mL and used to generate a standard curve spanning at least five concentration points. The limit of quantification (LOQ) was 1 ng/mL. The hexadeuterated internal standard (Toronto Research Chemicals Inc, Toronto, Canada; catalogue number C046502, 99.6% isotopic purity) was dissolved to 1 mg/mL in acetone and added at the same concentration to all standards and samples to compensate for possible matrix effects.
CAB encapsulation efficiency (%) in PACA NPs was calculated by the following formula:
4.4. In Vitro Cytotoxicity Studies
LP-9 normal human mesothelial cells (AG07086, Coriell Institute for Medical Research, Camden, NJ, USA) were cultivated in a humidified atmosphere at 5% CO2 and 37 °C in Medium 199 (M2154, Sigma-Aldrich (St. Louis, MO, USA)/MCDB110 (mixed 1 + 1). The MCDB110 medium was prepared in-house as follows: MCDB110 powder (15.3 g, US Biological, Salem, MA, USA) and sodium bicarbonate (2.2 g, Sigma-Aldrich, St. Louis, MO, USA) were dissolved in 900 mL distilled water. pH was adjusted to 7.4 by adding 2 M NaOH before the volume was brought to 1 L with distilled water. The solution was sterile filtered before use. The Medium 199/MCDB110 solution was supplemented with 1% (v/v) L-glutamine (G7513, Sigma-Aldrich, St. Louis, MO, USA), 1% (v/v) penicillin-streptomycin (15140122, Thermo Fisher Scientific, Waltham, MA, USA), 15% (v/v) fetal bovine serum (F7524, Sigma Aldrich, St. Louis, MO, USA), 10 ng/mL epidermal growth factor (E9644, Sigma-Aldrich, St. Louis, MO, USA) and 0.4 µg/mL hydrocortisone (H0888, Sigma-Aldrich, St. Louis, MO, USA).
For toxicity screening, LP-9 cells were seeded by adding 1875 cells (100 µL) per well in 96 well plates (165305, Thermo Fisher Scientific, Waltham, MA, USA). Following 24 h of incubation, empty PACA NPs or PACAlg were added at concentrations ranging from 0.0008 to 2 mg NPs/mL or 0.00072 to 0.18 mg NPs/mL, respectively. Staurosporine (S4400, Sigma-Aldrich, St. Louis, MO, USA) was included as positive control (0.0016 to 0.042 mg/mL). The plates were incubated for 48 or 72 h before viability was assessed by the addition of CellTiter-Glo 2.0 assay (Promega, Madison, WI, USA) (1 + 1). The luminescence signal was detected with a SpectraMax i3x multi-mode microplate reader after 10 min.
4.5. In Vivo Studies
All procedures and experiments involving mice were approved by the Norwegian Food Safety Authority (application ID #18209) and conducted according to the recommendations of the European Laboratory Animals Science Association and the ARRIVE guidelines [
40]. Female athymic nude foxn1
nu mice (6–8 weeks, 20–25g) were bred at the Department of Comparative Medicine, The Norwegian Radium Hospital, and kept in a specific pathogen free environment at constant temperature (22 ± 1 °C) and humidity (62 ± 5%) and with 15 air changes/hour and a 12-h light/dark cycle. Mice were purchased at 3 weeks, and then moved from the breeding room to the experimental room and allowed to acclimatize for 3–5 weeks until the start of the experiments. A maximum of nine mice were housed in each cage. Food and water were supplied ad libitum, and mice were given cardboard houses and paper to have nesting material and for environmental stimulation, as required by the Department of Comparative Medicine at the Norwegian Radium Hospital to improve the welfare of the mice [
41].
4.6. Biodistribution Study
For biodistribution experiments, PACAlg-CAB (6 mg CAB/kg, 500–600 µL) was injected intraperitoneally. Mice were euthanized after 1, 2, 3, 7, 14, and 21 days (two mice per time point) by cervical dislocation after a cardiac puncture to collect blood under 3% sevofluran anesthesia. Liver, spleen, peritoneum, adipose tissue, and a section of the parietal peritoneum, were excised from near the injection site and visible PACAlg-CAB microparticles were harvested. All samples were snap-frozen in liquid nitrogen and stored at −80 °C until further processing and LC-MS/MS analysis as described above. Blood was collected in EDTA tubes (Sarstedt Microvette® K3 EDTA, 500 µL, Sarstedt, Nümbrecht, Germany), mixed, and stored at −80 °C before LC-MS/MS analysis.
The tissue homogenization protocol published by Fusser, et al. 2019 [
18] was used to process the sample tissues and release the CAB content. A total of 1 mL of freshly prepared enzyme solution was added per 50 mg tissue and incubated at 37 °C for 48 h until the tissue was completely dissolved. Drug stability under these conditions was verified by including a free CAB control. All tissue homogenates and blood samples were diluted 10× in acetone added internal standard to extract the free drug and simultaneously precipitate sample proteins. The protein precipitates were then sedimented by centrifugation, and the supernatant was transferred to an HPLC vial for analysis. PACAlg-CAB microparticles were accurately weighed and transferred to Eppendorf tubes with 100 µL acetone added internal standard. All tubes were left for drug extraction by gentle rotation on a rotating mixer for 48 h at room temperature. The microparticles were then sedimented by centrifugation, and the supernatant was transferred to vials for analysis.
4.7. Treatment Efficacy Study
For treatment efficacy experiments, the PDX models PMCA-1 and PMCA-3 were used. The establishment of the models from patients with mucinous peritoneal metastases was previously described [
42]. Both models were established by implanting peritoneal tissue samples collected at the time of CRS-HIPEC. PMCA-1 was derived from a patient with a primary rectal carcinoma, while PMCA-3 was derived from a patient with high-grade mucinous cancer with signet cell differentiation. 200 µL of mucinous ascites were injected intraperitoneally. Treatment with 15 mg/kg CAB, PACAB, or PACAlg-CAB (500–600 µL per mouse) was initiated the following day to mimic the clinical situation after CRS with a low tumor load intraabdominally. CAB was dissolved to 40 mg/mL in Tween-80 (Sigma-Aldrich, St. Louis, MO, USA), and then in 10 mg/mL in 13% ethanol, before diluting in 0.9% NaCl to achieve the correct concentration. PACAB and PACAlg-CAB were diluted in 0.9% NaCl. Mice were randomly distributed to treatment groups of six mice. The mice were euthanized by cervical dislocation when they displayed a large distended abdomen or 100–102 days after treatment initiation if the mice did not develop a tumor. Two mice (PMCA-1, PACAlg-CAB, and PMCA-3, PACAB) were excluded from analyses due to death not occurring from tumor growth.
4.8. Statistical Analysis
Statistical analyses were performed using GraphPad Prism v7 and v9 (GraphPad Software, LaJolla, CA, USA). Student’s t-tests were used to compare differences between treatment groups. Survival curves (Kaplan–Meier plot) were compared using the Gehan-Breslow Wilcoxon test. p values < 0.05 were considered significant.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}