Abstract
Six new sesquiterpene aminoglycosides, trichaspside F (2) and cyclonerosides A–E (5–9), two new diterpene aminoglycosides, harzianosides A and B (10, 11), and three known sesquiterpenes, trichodermoside (1), cycloneran-3,7,10,11-tetraol (3), and cyclonerodiol (4), have been isolated from the n-butanol extract of Trichoderma sp. SCSIOW21 (Hypocreaceae), a deep-sea-sediment-derived fungus. The structures and relative configurations of the new compounds were determined using spectroscopic techniques and comparisons with those reported in the literature. The absolute configurations of the aglycone part of cyclonerosides A–E (5–9) were tentatively proposed based on optical rotation and biogenic considerations. Cyclonerosides A–E (5–9) represent the first glycosides of cyclonelane-type sesquiterpenes generated from Trichoderma. The NO-production-inhibitory activities were evaluated using macrophage RAW264.7 cells. Among the isolated compounds, trichaspside F (2) and cyclonerosides B–E (6–9) exhibited the strongest NO-production-inhibitory activities with IC50 values of 54.8, 50.7, 57.1, 42.0, and 48.0 µM, respectively, compared to the IC50 value of 30.8 µM for the positive control (quercetin). When tested for anti-fungal activities against several pathogenic fungi, none of the compounds exhibited significant activities at a concentration of 100 µM.
1. Introduction
Filamentous fungi have larger genomes and more biosynthetic gene clusters (BGCs) compared to bacteria, which leads to greater chemical diversity, making them one of the most important resources for natural product drug discovery [1]. However, with the intense study carried out over the last century, the high rediscovery rate from terrestrial fungi has seriously hindered new drug development from this resource. Research on natural products from marine fungi has received a great amount of research attention in recent years. A total of 346 compounds have been characterized from marine-sediment-derived fungi during 2005–2015 [2,3], in which almost half of the compounds (157) are reported to exhibit anti-microbial and anti-cancer activities. Further, 246 compounds have been reported from marine-sediment-derived fungi during 2016–2020 [4]. Among them, 12 compounds exhibit antiviral activity, 57 compounds have anti-microbial activity, and 62 compounds display cytotoxicity. In addition, 70 compounds have cytoprotective activity, including anti-infection, anti-oxidant, and neuroprotective activities, which demonstrates the diversity of the research being conducted on compound activity. However, anti-microbial and anti-cancer activities are still of significant interest in marine fungi natural products. Approximately 200 more compounds exhibiting anti-microbial and anti-cancer activities were reported in 2019–2022 from marine fungi [1]. Considering the best-selling drug lovastatin as an example, which contains hypolipidemic activity and was first discovered from a Penicillium species fungal strain, more bioassays should be prepared to screen for natural products in marine fungi to discover new active compounds with potential applications.
The number of Trichoderma spp. fungi isolated from nature was limited to dozens just a few decades ago. However, the introduction of molecular evolutionary approaches has led to the rapid expansion of the Trichoderma taxonomy, resulting in the discovery of ~50 new species each year. By July 2020, 361 Trichoderma species had been successfully cultivated and their DNA barcoded [5]. Trichoderma species are considered to be treasure troves of natural products. By 2008, 186 compounds had been identified from Trichoderma [6], while 203 compounds were characterized between 2009 and 2020, including terpenoids, cyclopeptides, diketopiperazines, alkaloids, and polyketides [7]. Among filamentous fungi, Trichoderma species are some of the dominant producers of terpenoids. However, glycosylated sesquiterpenes and diterpenes have been rarely reported in the literature [8]. To the best of our knowledge, three trichothecene glycosides, namely trichodermarins L–N, have been discovered in marine algicolous Trichoderma brevicompactum A-DL-9-2 [9], while one bisabolane acetamido glycoside, trichodermoside, has been isolated from marine-derived Trichoderma sp. PT2 [10]. Three bisabolane acetamido glycosides, trichaspsides C–E, have been isolated from Trichoderma asperellum A-YMD-9-2, a marine algal endophytic fungus [11], and trichosordarin A, a diterpene glycoside, has been isolated from the marine-derived Trichoderma harzianum R5 fungus [12].
Anti-microbial, anti-microalgae, anti-cancer, and phytotoxic activities have been reported for sesquiterpenes and diterpenes isolated from Trichoderma species [7]. Cyclonerane sesquiterpenes also exhibit nematocidal activity [13]. However, the effects of sugar moieties on the activities of these compounds remain under debate. The sugar moiety in bisabolene-type sesquiterpenes has no effect on their growth-inhibition activities against marine phytoplankton species [11], and glycosylation of trichothecene sesquiterpenes appears to reduce their anti-fungal and anti-microalgae activities [9]. In contrast, the amino sugar moiety in bisabolane sesquiterpenes is indispensable for their activities against several aquatic pathogenic bacteria [14]. Trichodermoside, a bisabolane sesquiterpene glycoside, has been shown to weakly inhibit the growth of human HeLa cells [10].
During our research on anti-inflammatory natural products obtained from deep marine fungi [15,16,17,18], we found that the n-butanol (n-BuOH) extract of the Trichoderma species SCSIOW21 inhibits nitric oxide (NO) production in RAW 267.4 cells stimulated by LPS. In the present study, we have characterized six new aminoglycoside sesquiterpenes, trichaspside F (2) and cyclonerosides A–E (5–9), two new aminoglycoside diterpenes, namely harzianosides A and B (10, 11), and three known sesquiterpenes, trichodermoside (1), cycloneran-3,7,10,11-tetraol (3), and cyclonerodiol (4), from the fungal culture (Figure 1). The NO-production-inhibitory activities and anti-fungal activities of these compounds were studied.
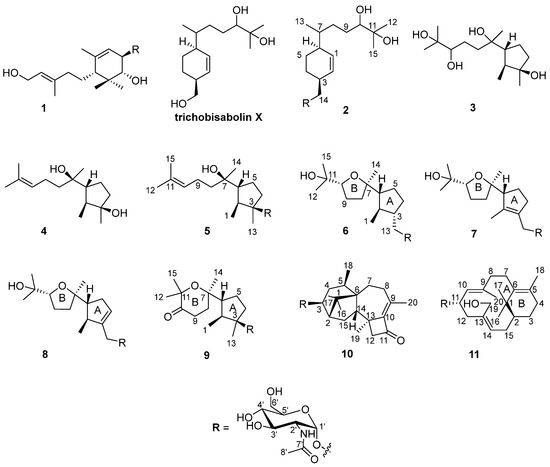
Figure 1.
Structures of compounds 1–11.
2. Results and Discussion
The fungal strain was statically cultivated in rice broth containing 3% sea salt and then extracted with n-BuOH. The extract was subjected to silica gel column chromatography, followed by HPLC on an ODS C18 column to obtain 11 compounds (Figure 1).
Compound 2 was isolated as a colorless gum and its molecular formula was determined to be C23H41NO8 using HRESIMS. The ESI-MS fragment-ion peak observed at m/z 204 was indicative of the presence of an acetamido-sugar moiety [9,11,14]. The 1H, 13C, DEPT, and HSQC NMR spectra show four sp3 methyl, six sp3 methylene, nine sp3 methine, and two sp2 methine signals, as well as peaks corresponding to one sp3 quaternary carbon and one ketone carbonyl carbon (Table 1). Apart from the additional 1H NMR signals observed at δH [4.67, d (3.0) H-1’], [3.62, m H-2’], [3.45, m H-3’], [3.12, m H-4’], [3.38, m H-5’], [3.59, m H-6’a], [3.48, m H-6’b], [1.83, s Me-8’], and [7.68, d (8.0) -NH], and the 13C NMR signals at δC [97.1, C-1’], [54.0, C-2’], [70.6, C-3’], [70.7, C-4’], [72.8, C-5’], [60.7, C-6’], [169.4, C-7’], and [22.5, Me-8’], the NMR data were almost identical to those reported for trichobisabolin X [19], a bisabolane-type sesquiterpene, which suggests the presence of an acetamido-substituted sugar residue [9,11,14]. Unfortunately, most of the proton signals in the sugar region overlap in DMSO-d6 (Table 1), and consequently the coupling constants could not be determined. The 1H and 13C NMR data were re-acquired in MeOD-d4. The chemical shifts and proton coupling constants in the sugar region [δH 4.81, d (3.0 Hz) H-1’; δH 3.86, dd (10.0, 4.0 Hz) H-2’; δH 3.62, dd (10.0, 9.0 Hz) H-3’; δH 3.36, dd (10.0, 9.0 Hz) H-4’, δH 3.58, m H-5’; δH 3.80, dd (12.0, 2.0 Hz) H-6’a; δH 3.69, dd (12.0, 5.0 Hz) H-6’b; δH 1.99, s Me-8’] are almost identical to the 2-acetamido-2-deoxy-α-glucopyranosyl signals in trichaspsides A–E (Table 1) [11,14]. NOE correlations observed for H-2’/H-4’ and H-3’/H-5’ further confirmed the relative configuration of the glucopyranoside. The small anomeric-proton coupling constant (3.0 Hz) suggests that the sugar linkage has an α-configuration. The aglycone of 2 was further analyzed using 1H-1H correlation spectroscopy (COSY) and heteronuclear multiple bond correlation (HMBC) (Figure 2). The 1H-1H COSY correlations connect C-1 to C-10 and the C13 methyl group (Figure 2). The two singlet methyl groups are not correlated in the COSY spectrum. The two methyl groups (δH 0.98 s and 1.03 s) show HMBC correlations with C-10 at δC 77.4 and another quaternary carbon at δC 71.6. One additional hydroxy group at δH 4.03 also exhibits a HMBC correlation with this quaternary carbon. Therefore, the presence of an isopropanol moiety is proposed to be linked to C-10. The aglycone moiety was determined to be the same as that observed in trichobisabolin X upon further comparison of the NMR spectra (Table 1 and Figure 2) [14]. The sugar residue was determined to be attached to C-14 by the mutual HMBC correlations observed between H-1’ and C-14, and H2-14 and C-1’ (Figure 2). H-3/H-5a and H-5a/H-6 ROESY correlations and the identical NMR data obtained for the aglycone suggest the same relative configuration to that observed in trichobisabolin X (Table 1 and Figure 3). Since three bisabolane acetamido glycosides, trichaspsides C–E, have been previously reported [11,14], we assigned compound 2 as trichaspside F.

Table 1.
1H and 13C NMR data (600 and 150 MHz, respectively) obtained for compound 2.
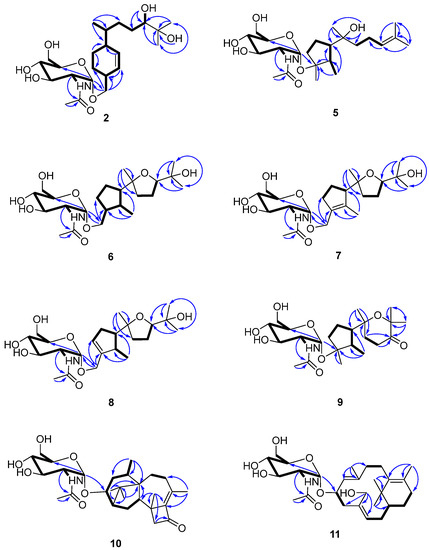
Figure 2.
Key 2D NMR spectroscopic correlations observed for compounds 2 and 5–11.
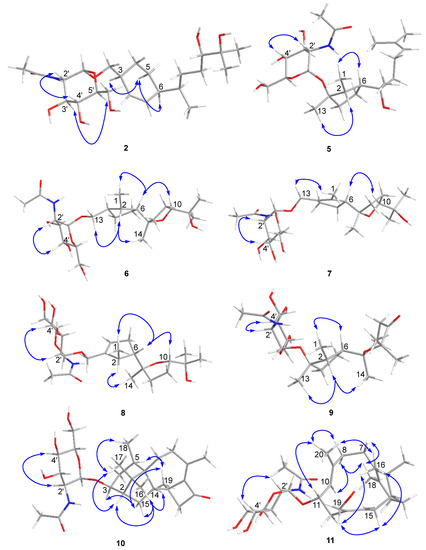
Figure 3.
Key NOE correlations observed for compounds 2 and 5–11.
Compounds 5–11 were isolated as colorless gums. Their ESI-MS spectra exhibit representative base peaks at m/z 204, which imply the presence of an acetamido-sugar moiety in each structure [9,11,14]. The sugar region exhibits 1H and 13C NMR data that are almost identical to those reported for trichaspside F (2), which reveals the presence of a 2-acetamido-2-deoxy-α-glucopyranosyl moiety in each of these molecules (Table 2, Table 3 and Table 4). The small coupling constant observed for each aromatic proton (3.0 Hz) suggests all of the sugar linkages have an α-configuration (Table 2 and Table 4).
Table 2.
1H NMR data (600 MHz) obtained for compounds 5–9a.
Table 3.
13C NMR data (150 MHz) obtained for compounds 5–9a.
Table 4.
1H and 13C NMR data (600 and 150 MHz, respectively) obtained for compounds 10 and 11 a.
The molecular formula of compound 5 was determined to be C23H41NO7 using HRESIMS. The 1H-1H COSY analysis shows that the methine at H-10 is connected to the methylene at H2-9, and H2-9 is connected with another methylene at H2-8. The 1-Me methyl group is connected to the methine at H-2 and extends from H-2 to H-6, H2-4, and H2-5 (Figure 2). From the HMBC spectrum, two methyl groups (δH 1.56 s, 1.63 s) are linked to the quaternary carbon at C-11 (δC 130.1). 14-Me (δH 1.00 s) exhibits HMBC correlations with C-6 and C-7. 7-OH (δH 3.89) shows HMBC correlations with C-7 and C-9. These correlations connect the partial structure between C-6 and C-9 (Figure 2). Both methyl groups at 1-Me and 13-Me show HMBC correlations with the quaternary carbon at C-3 (δC 86.3) (Figure 2). Detailed 1H-1H COSY and HMBC analyses were then conducted to establish the structure of the aglycone (Figure 2 and Figures S10 and S18). With the exception of the sugar region, compound 5 exhibits 1H and 13C NMR data that are almost identical to those reported for cyclonerodiol (4), a cyclonerane-type sesquiterpene (Table 2 and Table 3) [19]. HMBC correlations between H-1’ and C-3 establish that the sugar moiety is linked to C-3 (Figure 2), and the ROESY correlations observed for Me-1/H-6 and Me-13/H-2 reveal that the relative configurations of the five-membered ring are identical to those of cyclonerodiol (4) (Figure 3) [20]. Unfortunately, the configuration at C-7 could not be determined. Thus, compound 5 is identified to be 3-O-cyclonerodiol-2-acetamido-2-deoxy-α-glucopyranoside (Figure 1). As it is the first glycosylated cyclonerane-type sesquiterpene discovered to date, we have assigned compound 5 as cycloneroside A. Cyclonerodiol (4) was obtained in this study with a negative optical rotation of −18, which is similar to the reported −20 of [21]. Considering that cycloneroside A (5) and cyclonerodiol (4) possess the same sesquiterpene carbon skeleton and presumably the same biogenetic pathway (Figure 4), the absolute configurations of the aglycone part of cycloneroside A (5) were thus assigned as that of cyclonerodiol (4) based on the biogenic consideration as 2S, 3R, 6R, 7R [21,22,23]. The absolute configurations of the sugar moiety were not able to be assigned due to the lack of materials.
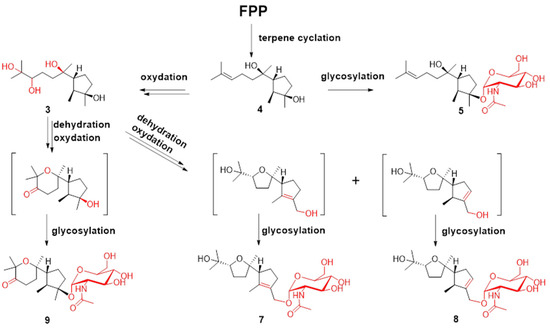
Figure 4.
Plausible biosynthesis routes for compounds 3–5 and 7–9.
The molecular formula of compound 6 was determined to be C23H41NO8 using HRESIMS. Apart from the sugar region and the additional signals observed for an oxygenated methylene group [δH 3.54 m, 3.15 m; δC 68.9], the sp3 methine signal [δH 2.11 m, H-3; δC 43.2], and lack of signals corresponding to the methyl group at C-13 and oxygenated quaternary carbon at C-3 (Table 2 and Table 3), compound 6 exhibits NMR data that highly resemble those reported for isoepicyclonerodiol oxide [19]. The additional oxygenated methylene and methine groups are located at C-13 and C-3, respectively, based on the 1H-1H COSY correlations observed between H-13 and H-3, and H-3, H-2, and H-4, and the HMBC correlations observed between H-13 and C-3 and C-4, and between 1-Me and C-3 (Figure 2). The 2D NMR spectra were then carefully analyzed, which confirmed the presence of an aglycone, as shown in Figure 2. The sugar moiety is linked at C-13 according to the HMBC correlations observed between H-1’ and C-13, and H-13 and C-1’ (Figure 2 and Figures S19–S26). Further, the ROESY correlations observed between Me-1/H-6 and Me-13/H-2 indicate that ring A has the same relative configuration as compound 5. The ROESY correlations observed for H-6/H-10 and H-2/Me-14 (Figure 3), and the almost identical NMR data obtained for ring B suggest the same relative configuration as isoepicyclonerodiol oxide [19]. Thus, compound 6 was identified to be a 13-O-isoepicyclonerodiol oxide-2-acetamido-2-deoxy-α-glucopyranoside, which we assigned as cycloneroside B. The absolute configurations of the aglycone part of cycloneroside B (6) were assigned based on the biogenic consideration as 2S, 3S, 6R, 7R, 10R.
The molecular formula of compound 7 was determined to be C23H39NO8 using HRESIMS. The IR absorption bands of amide I at 1639 cm−1 (C=O stretching vibration) and amide II at 1555 cm−1 (N-H bending vibration) indicated the presence of a secondary amide group [24]. With the exception of the sugar region, and the lack of the sp3 methine signals at C-2 and C-3 [δH 2.22, m, δC 35.5; δH 2.11, m, δC 43.2] and two additional sp2 olefinic quaternary carbons [δC 134.8; δC 137.5] (Table 2 and Table 3), the 1H and 13C NMR data obtained for 7 resemble those of 6 (Figures S27–S34). The Me-1 proton signal of compound 7 (δH 1.66) is down-field-shifted when compared to that of 6 [δH 0.68, d (7.0 Hz)] and appears as a singlet. The methylene signals at position 13 are also low-field shifted (δH 3.98 and 4.10) relative to those observed for 6 (δH 3.54, m; δH 3.15, m) with a geminal coupling observed (J = 12 Hz), which implies the presence of two olefinic carbons at C-2 and C-3 (Table 2). These positions were further validated using the HMBC correlations observed between 1-Me and H-13 and these two olefinic carbons (Figure 2). The sugar moiety is linked at C-13 according to the HMBC correlations observed between H-1’ and C-13, and H-13 and C-1’ (Figure 2). The ROESY correlation observed between Me-1 and H-13 suggests the presence of a trans double bond. The almost identical NMR data obtained for ring B and the H-6/H-10 ROESY correlation suggest the same relative configuration as that observed for compound 6 (Table 2 and Figure 3 and Figures S35–S43). Hence, the structure of compound 7 is elucidated (Figure 1) and assigned as cycloneroside C. The absolute configurations of the aglycone part of cycloneroside C (7) re assigned based on the biogenic consideration as 6R, 7R, 10R.
The molecular formula of compound 8 was determined to be C23H39NO8 using HRESIMS. The IR absorption bands at 1653 and 1555 cm−1 indicated the presence of a secondary amide group. With the exception of one additional sp2 methine [δH 5.54 (br s); δC 126.8, CH] and an sp3 methine [δH 2.46 m; δC 41.0, CH], the lack of one sp2 quaternary carbon at C-2 and one sp3 methylene group at C-4, which implies that the double bond is positioned between C-3 and C-4, compound 8 exhibits 1H and 13C NMR data that resemble those of 7 (Table 2 and Table 3). The 1H-1H COSY correlations observed between H-4 and H-5 (δH 2.32, m; δH 2.06, m) and H-2 and 1-Me [δH 1.06, d (7.0)] (Figure 3) confirm the position of the new double bond. The HMBC correlations between H-13 and C-3 and C-4, and between 1-Me and C-2 and C-3 also confirm this assignment (Figure 2). The sugar moiety is linked at C-13 according to the HMBC correlations observed between H-1’ and C-13, and H-13 and C-1’ (Figure 2). The ROESY correlation observed for Me-1/H-6 suggests that these protons are directed toward the same face of the molecule. The other ROESY correlations and NMR data obtained for ring B are almost identical to those observed for compound 6, suggesting they have the same relative configuration (Table 2 and Figure 3). We assigned compound 8 as cycloneroside D. The absolute configurations of the aglycone part of cycloneroside D (8) were assigned based on the biogenic consideration as 2S, 6R, 10R.
The molecular formula of 9 was determined to be C23H39NO8 using HRESIMS. The 1H and 13C NMR data obtained for the aglycone are very similar to those of 3,7,11-trihydroxycycloneran-10-one [25]. Detailed 1H COSY and HMBC analysis confirms the skeleton (Figure 2 and Figures S44–S51); however, MS reveals an m/z difference of 18, which suggests dehydration between the two hydroxyl groups at C-7 and C-11 (Table 2 and Table 3). The coupling patterns observed for H-8 [δH 2.04 (13.0, 10.0, 6.0); δH 1.88 (13.0, 6.0, 5.0)] and H-9 [δH 2.53 (17.0, 10.0, 6.0); δH 2.42 (17.0, 6.0, 5.0)] also suggest a relatively rigid tetrahydropyran for ring B instead of a linear structure [25]. The sugar moiety is linked to C-3 according to the HMBC correlation observed between H-1’ and C-3 (Figure 2). The almost identical NMR data obtained for ring A and the ROESY correlations observed for Me-1/H-6 and Me-13/H-2 suggest that compound 9 has the same relative configuration to that of ring A in compound 5 (Table 2 and Figure 3). The ROESY correlation observed at H-2/Me-14 indicates that the two groups are directed toward the same side of the molecule (Figure 3). Thus, the structure of 9 is elucidated (Figure 1) and assigned as cycloneroside E. The absolute configurations of the aglycone part of cycloneroside E (9) was assigned based on the biogenic consideration as 2S, 3R, 6R, 10R.
The molecular formula of compound 10 was deduced to be C28H43NO7 using HRESIMS. The IR absorption bands at 1742, 1632, and 1563 cm−1 indicated the presence of a carbonyl and a secondary amide group. With the exception of the additional signals corresponding to the acetamido-sugar moiety, the aglycone exhibits 1H and 13C NMR data consisting of one ketone (C-11), two sp2 quaternary carbons (C-9 and C-10), and 17 sp3 carbons including five methyl (C-16, C-17, C-18, C-19, and C-20), five methylene (C-4, C-7, C-8, C-12, and C-15), one oxygenated methine (C-3), three methines (C-2, C-5, and C-14), and three non-protonated carbons (C-1, C-6, and C-13) by HSQC and DEPT analyses (Table 4). Careful examination of the NMR data, particularly 1H-1H COSY and HMBC correlations (Figure 2), confirmed the presence of a harziane-type tetracyclic scaffold consisting of a 6/5/7/4-fused tetra-cyclic ring system. The NMR data resemble those of 3S-hydroxyharzianone, a harziane-type diterpene (Table 4 and Figures S52–S58) [26,27], except for the additional amino sugar moiety. The sugar moiety is attached to C-3 according to the HMBC correlation observed between H-1’ and C-3 (Figure 2), and the relative configurations of compound 10 are fully established using ROESY, which exhibit correlations for Me-18/Me-17, Me-17/H-2, Me-16/H-2, Me-16/H-14, and Me-16/H-15a. This further suggests that these protons point toward the same side of the molecule, whereas the ROESY correlations observed for H-3/H-15b, Me-19/ H-15b, and Me-19/H-5 indicate that these protons are placed on the other side of the molecule (Figure 3). This compound is assigned as harzianoside A.
The molecular formula of compound 11 was determined to be C28H45NO7 using HRESIMS. With the exception of the sugar moiety, the aglycone exhibits 1H and 13C NMR data comprised six sp2 carbons including four quaternary double bond carbons (C-5, C-6, C-9, and C-13) and two methines (C-10 and C-14); and 14 sp3 carbons including one quaternary carbon (C-1), one oxygenated methine (C-11), one oxygenated methylene (C-19), one methine (C-2), six methylene (C-3, C-4, C-7, C-8, C-12, and C-15), and four methyl (C-16, C-17, C-18, and C-20) groups by HSQC and DEPT analyses (Table 4). Detailed 1H-1H COSY and HMBC analyses reveal the bicyclic core structure of 11, which contains cyclohexene and cyclododecadiene rings (Figure 2 and Figures S59–S65). The aglycone exhibits 1H and 13C NMR data that are almost identical to those of 11R-methoxy-5,9,13-proharzitrien-19-ol (Table 4) [28]. The sugar moiety is linked to C-11 according to the HMBC correlation observed for H-1’/C-11 (Figure 2). The Me-18/H-7, Me-20/H-11, and H-15/H-19 ROESY correlations suggest the presence of 5-6-cis, 9-10-trans, and 13-14-cis double bonds. The NOE correlations observed for Me-20/Me-16, Me-20/H-8b, M-20/H-11, Me-16/H-15, and Me-16/H-19 suggest that these protons are directed to the same side of the molecule, whereas the NOE correlations for Me-18/H-8a and H-8a/H-10 indicate that these protons point to the other side of the molecule (Figure 3). Therefore, the structure of compound 11 is elucidated (Figure 1) and assigned as harzianoside B.
The structures of the three known sesquiterpenes, trichodermoside (1) [10], cycloneran-3,7,10,11-tetraol (3) [25], and cyclonerodiol (4) [20], were determined by comparison with the data reported in the literature.
Terpenes are biosynthesized from C-5 building blocks composed of isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). Prenyl transferase assembles IPP and DMAPP into farnesyl diphosphate (FPP), which is the universal precursor for the sesquiterpenes. FPP is cyclized by various terpene cyclases to form different types of sesquiterpenes. The post-tailoring steps further diversify these structures [29,30]. The plausible biosynthesis routes to cyclonerane-type sesquiterpenes in this study are summarized in Figure 4. Cyclonerodiol (4) has been demonstrated to be the direct product of FPP cyclization by terpene cyclase [22]. The reactions observed in the post tailoring steps may include complex oxidation, dehydration, and glycosylation (Figure 4). Notably, the N-acetylaminosugar usually needs to undergo additional transamination and acetylation reactions prior to the glycosylation reaction [31,32].
The abilities of all of the isolated compounds to inhibit NO production were examined using macrophage RAW 264.7 cells stimulated by LPS [18]. Figure 5 shows that all of the compounds inhibit NO production in a dose-dependent manner. None of them exhibit cytotoxicity at the maximum tested concentration (100 µM) (Figure 6). A comparison of the data obtained for cyclonerodiol (4) and cycloneroside A (5) suggests that the sugar moiety appears to have little impact on the activity of this type of compound. Trichaspside F (2) and cyclonerosides B–E (6–9) exhibit the strongest abilities to inhibit NO production with IC50 values of 54.8, 50.7, 57.1, 42.0, and 48.0 µM, respectively. The other compounds in this study show low activity with IC50 values ~100 µM. Our structure–activity relationship analyses reveal that the tetrahydropyran or tetrahydrofuran ring is essential toward improving the activities of cyclonerosides B–E (6–9) when compared with those of cycloneroside A (5), which contains a linear prenyl chain on the right hand side of the structure. The positive control (quercetin) exhibits an NO-production-inhibitory effect with an IC50 value of 30.8 µM.
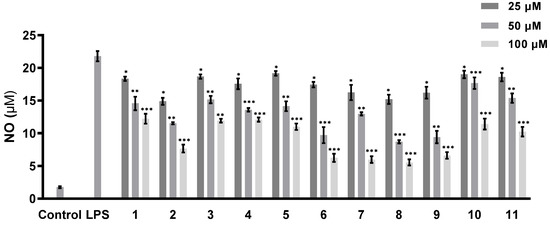
Figure 5.
NO-production-inhibitory activities of compounds 1–11. The values represent the mean ± SEM of three independent experiments. * p < 0.05; ** p < 0.01; *** p < 0.001.
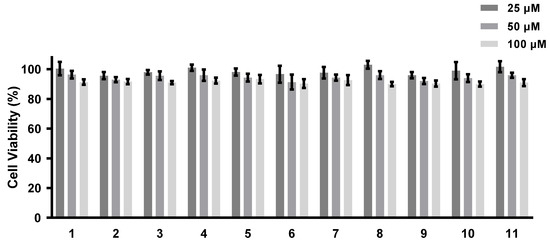
Figure 6.
Cell viability of compounds 1–11. The values represent the mean ± SEM of three independent experiments.
Inflammatory responses are known to be deeply associated with the pathogenesis of various diseases, such as diabetes, cardiovascular diseases, obesity, arthritis, stroke, and cancer [17]. The over-expression of cellular transduction molecules, such as NO, histamine, and other pro-inflammatory cytokines, leads to chronic inflammation, which eventually induces inflammation-related diseases. Chemical inhibitors of cell signal transduction that can inhibit the excessive release of these harmful cytokines should be useful as potential drug candidates [17,33]. There has been a large number of reports on natural products with NO-production-inhibitory activity isolated from plants, most of which are phenolic-type components [33]. To the best of our knowledge, there have been no reports on the NO-production-inhibitory activities of sesquiterpene aminoglycosides. These compounds do not show any cytotoxicity at the highest test concentration, and further structural modification of these compounds may lead to new, potent, and safe anti-inflammatory drugs.
The anti-fungal activities were also tested against plant pathogenic fungi including Helminthosporium maydis, Gibberella sanbinetti, and Penicillium digitatum. None of the compounds exhibit any obvious activity at a concentration of 100 µg/mL.
3. Materials and Methods
3.1. General Experimental Procedures
NMR spectroscopy was performed using a Bruker ASCEND 600 MHz NMR spectrometer equipped with a CryoProbe (Bruker Biospin GmbH, Rheinstetten, Germany). Optical rotations were measured on an Anton Paar MCP-100 polarimeter (Anton Paar GmbH, Graz, Austria) and the HRESIMS spectra were acquired on a MaXis quadrupole-time-of-flight mass spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany). Silica gel (200–300 mesh, Qingdao Haiyang Chemical, Qingdao, China) and YMC Gel ODS-A (150 µM, YMC Co., Ltd., Kyoto, Japan) were used for column chromatography and silica gel 60 F254 and RP-18 F254 thin-layer chromatography (TLC) plates (Merck Millipore Co., Darmstadt, Germany) were used for TLC. HPLC was performed using a Shimadzu LC-16P system (Shimadzu Co., Tokyo, Japan) equipped with a YMC-Pack ODS-A C18 column (20 × 250 mm, 5 µm; YMC Co., Ltd., Kyoto, Japan). Analytical- and HPLC-grade reagents (Macklin Co., Shanghai, China) were used for the isolation procedures.
3.2. Fungal Strain and Fermentation
The Trichoderma sp. SCSIOW21 fungal strain was collected from deep-sea sediments and deposited at the Laboratory of Microbial Natural Products, Shenzhen University, China. The fungal strain was identified as Trichoderma species according to its morphological characteristics and ITS gene sequence (OP854922). The fungal strain was cultivated statically for 30 d in rice broth containing 3% sea salt. The culture was extracted using an equal volume of n-BuOH.
3.3. Isolation Procedure
The n-BuOH extract was subjected to silica gel column chromatography with gradient elution using CH2Cl2/MeOH/water (100:0:0, 50:1:0, 20:1:0, 10:1:0, 5:1:0.1, 3:1:0.1, 1:1:0.1, and 0:0:100, v/v/v, 2.0 L of each) to yield eight fractions (A–H). Fraction B was separated using a medium-pressure YMC Gel ODS-A (150 µM) column with a MeOH/water gradient (50–100% MeOH) to give five sub-fractions. Subfraction 3 was separated using a YMC-Pack ODS-A C18 column [acetonitrile (ACN)/water, 50:50 v/v] to give compound 4 (tR 19.1 min, 10 mL/min, 0.7 mg). Fraction E was separated using a medium-pressure YMC Gel ODS-A (150 µM) column with a MeOH/water gradient (10–100% MeOH) to give 18 sub-fractions. Subfraction 9 was separated using a YMC-Pack ODS-A C18 column (ACN/water, 12:88 v/v) to yield compound 3 (tR 19.1 min, 10 mL/min, 0.7 mg). Subfraction 15 was separated using HPLC on a YMC-Pack ODS-A C18 column (ACN/water, 23:77 v/v) to yield compound 1 (tR 24.7 min, 10 mL/min, 1.9 mg). Subfraction 16 was separated on the YMC-Pack ODS-A C18 column (ACN/water, 25:75) to give compounds 2, 7, 8, and 9 (tR2 20.7 min, 10 mL/min, 1.4 mg; tR7 24.6 min, 10 mL/min, 3.1 mg; tR8 26.1 min, 10 mL/min, 2.8 mg; tR9 31.0 min, 10 mL/min, 2.8 mg). Subfraction 17 was separated using a YMC-Pack ODS-A C18 column (ACN-Water, 33:67 v/v) to yield compounds 5, 6, and 10 (tR5 16.5 min, 10 mL/min, 0.6 mg; tR6 22.0 min, 10 mL/min, 0.6 mg; tR10 33.5 min, 10 mL/min, 2.0 mg). Subfraction 18 was separated on the YMC-Pack ODS-A C18 column (ACN/water, 40:60 v/v) to yield compound 11 (tR 31.5 min, 10 mL/min, 1.9 mg).
3.4. Spectral Data
Trichaspside F (2): Colorless gum;
+ 32 (c 0.17, MeOH); 1H and 13C NMR data (DMSO-d6, 600 and 150 MHz, respectively), see Table 1; HRESIMS m/z: 482.2744 [M + Na]+ (calcd. for C23H41NNaO8, 482.2730).
Cycloneroside A (5): Colorless gum; + 74 (c 0.14, MeOH); 1H and 13C NMR data (DMSO-d6, 600 and 150 MHz, respectively), see Table 2 and Table 3; HRESIMS m/z: 466.2769 [M + Na]+ (calcd. for C23H41NNaO7, 466.2781).
Cycloneroside B (6): Colorless gum; + 28 (c 0.21, MeOH); 1H and 13C NMR data (DMSO-d6, 600 and 150 MHz, respectively), see Table 2 and Table 3; HRESIMS m/z: 482.2743, [M + Na]+ (calcd. for C23H41NNaO8, 482.2730).
Cycloneroside C (7): Colorless gum; + 104 (c 0.26, MeOH); IR (KBr) vmax 3355, 2970, 1639, 1555, 1437, 1377, 1317, 1022, 950 cm−1; 1H and 13C NMR data (DMSO-d6, 600 and 150 MHz, respectively), see Table 2 and Table 3; HRESIMS m/z: 480.2581 [M + Na]+ (calcd. for C23H39NNaO8, 480.2573).
Cycloneroside D (8): Colorless gum; + 20 (c 0.5, MeOH); IR (KBr) vmax 3348, 2927, 1653, 1551, 1443, 1373, 1331, 1013 cm−1; 1H and 13C NMR data (DMSO-d6, 600 and 150 MHz, respectively), see Table 2 and Table 3; HRESIMS m/z: 480.2576 [M + Na]+ (calcd. for C23H39NNaO8, 480.2573).
Cycloneroside E (9): Colorless gum; + 56 (c 0.14, MeOH); 1H and 13C NMR data (DMSO-d6, 600 and 150 MHz, respectively), see Table 2 and Table 3; HRESIMS m/z: 480.2580, [M + Na]+ (calcd. for C23H39NNaO8, 480.2573).
Harzianoside A (10): Colorless gum; + 91 (c 0.81, MeOH); IR (KBr) vmax 3402, 2929, 1742, 1632, 1563, 1436, 1379, 1327, 1046, 972 cm−1; 1H and 13C NMR data (DMSO-d6, 600 and 150 MHz, respectively), see Table 4; HRESIMS m/z: 506.3117 [M + H]+ (calcd. for C28H44NO7, 506.3118).
Harzianoside B (11): Colorless gum; + 31 (c 0.15, MeOH); 1H and 13C NMR data (DMSO-d6, 600 and 150 MHz, respectively), see Table 4; HRESIMS m/z: 530.3084 [M + Na]+ (calcd. for C28H45NNaO7, 530.3094).
3.5. NO-Production-Inhibitory Activity
NO inhibition was investigated using LPS-stimulated RAW 264.7 cells [18]. Briefly, RAW264.7 cells were plated into a 96-well plate at a density of 1 × 105 cells/well. The tested samples were simultaneously added with LPS stimulation at final concentrations of 25, 50, and 100 μM. The cells were cultivated for 24 h and the supernatant tested for NO using the Griess reagent. Quercetin was used as a positive control with an IC50 value of 30.8 µM. Cells incubated for 4 h were subjected to an MTT assay [18].
3.6. Anti-fungal Activity
The mycelial-growth-inhibition effects were tested based on an assay protocol used in a previous publication [23].
4. Conclusions
Glycosylated terpenoids from the Trichoderma species of filamentous fungi have rarely been reported. In this study, eleven terpenes, including six new sesquiterpene and two new diterpene aminoglycosides, were isolated and characterized from a deep-sea-sediment-derived fungus, Trichoderma sp. SCSIOW21. Cyclonerosides A–E (5–9) represent the first glycosylated cyclonelane-type sesquiterpenes isolated from Trichoderma, which greatly increase the chemo-diversity of this species. The NO-production-inhibitory activities were tested for each of the isolated compounds. Trichaspside F (2) and cyclonerosides B–E (6–9) exhibit the strongest abilities to inhibit NO production with IC50 values of 54.8, 50.7, 57.1, 42.0, and 48.0 µM, respectively. Further studies on the structure–activity relationship and mechanism of action of these compounds may contribute to the development of new anti-inflammatory drugs.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/md21010007/s1, Figures S1–S65: NMR and MS spectra of compounds 2, and 5–11.
Author Contributions
The contributions from each of the respective authors are as follows: H.L. contributed to the fermentation, extraction, structural elucidation, and manuscript preparation; X.L. contributed to the fermentation, extraction, and isolation; Z.H. contributed to the manuscript revision; L.W. contributed to the experimental design, manuscript preparation, supervision, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key Research and Development Project with grant number 2018YFA0902504.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Materials.
Acknowledgments
The authors thank the Instrumentation Analysis Center of Shenzhen University and the College of Life Sciences and Oceanography for the NMR spectroscopy and MS data measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Giddings, L.A.; Newman, D.J. Extremophilic Fungi from Marine Environments: Underexplored Sources of Antitumor, Anti-Infective and Other Biologically Active Agents. Mar. Drugs 2022, 20, 62. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef]
- Jin, L.; Quan, C.; Hou, X.; Fan, S. Potential Pharmacological Resources: Natural Bioactive Compounds from Marine-Derived Fungi. Mar. Drugs 2016, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, A.N.; Girich, E.V.; Yurchenko, E.A. Metabolites of Marine Sediment-Derived Fungi: Actual Trends of Biological Activity Studies. Mar. Drugs 2021, 19, 88. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Druzhinina, I.S. In honor of John Bissett: Authoritative guidelines on molecular identification of Trichoderma. Fungal. Divers. 2021, 107, 1–69. [Google Scholar] [CrossRef]
- Reino, J.L.; Guerrero, R.F.; Hernández-Galán, R.; Collado, I.G. Secondary metabolites from species of the biocontrol agent Trichoderma. Phytochem. Rev. 2008, 7, 89–123. [Google Scholar] [CrossRef]
- Zhang, J.-L.; Tang, W.-L.; Huang, Q.-R.; Li, Y.-Z.; Wei, M.-L.; Jiang, L.-L.; Liu, C.; Yu, X.; Zhu, H.-W.; Chen, G.-Z.; et al. Trichoderma: A Treasure House of Structurally Diverse Secondary Metabolites with Medicinal Importance. Front. Microbiol. 2021, 12, 723828. [Google Scholar] [CrossRef]
- Jiang, M.; Wu, Z.; Guo, H.; Liu, L.; Chen, S. A Review of Terpenes from Marine-Derived Fungi: 2015–2019. Mar. Drugs 2020, 18, 321. [Google Scholar] [CrossRef]
- Shi, Z.-Z.; Liu, X.-H.; Li, X.-N.; Ji, N.-Y. Antifungal and Antimicroalgal Trichothecene Sesquiterpenes from the Marine Algicolous Fungus Trichoderma brevicompactum A-DL-9-2. J. Agric. Food Chem. 2020, 68, 15440–15448. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Lu, C.; Zheng, Z.; Shen, Y. A novel sesquiterpene glucoside from Trichoderma sp. PT2. Nat. Prod. Res. Dev. 2010, 22, 544–547. [Google Scholar]
- Song, Y.-P.; Miao, F.-P.; Liu, X.-H.; Yin, X.-L.; Ji, N.-Y. Seven chromanoid norbisabolane derivatives from the marine-alga-endophytic fungus Trichoderma asperellum A-YMD-9-2. Fitoterapia 2019, 135, 107–113. [Google Scholar] [CrossRef]
- Liang, X.-R.; Ma, X.-Y.; Ji, N.-Y. Trichosordarin A, a norditerpene glycoside from the marine-derived fungus Trichoderma harzianum R5. Nat. Prod. Res. 2020, 34, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Du, F.Y.; Ju, G.L.; Xiao, L.; Zhou, Y.M.; Wu, X. Sesquiterpenes and Cyclodepsipeptides from Marine-Derived Fungus Trichoderma longibrachiatum and Their Antagonistic Activities against Soil-borne Pathogens. Mar. Drugs 2020, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.P.; Liu, X.H.; Shi, Z.Z.; Miao, F.P.; Fang, S.T.; Ji, N.Y. Bisabolane, cyclonerane, and harziane derivatives from the marine-alga-endophytic fungus Trichoderma asperellum cf44-2. Phytochemistry 2018, 152, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; He, J.; Wu, Y.; Du, N.; Li, X.; Ju, J.; Hu, Z.; Umezawa, K.; Wang, L. Isolation and Characterization of New Anti-Inflammatory and Antioxidant Components from Deep Marine-Derived Fungus Myrothecium SP. Bzo-l062. Mar. Drugs 2020, 18, 597. [Google Scholar] [CrossRef]
- Li, H.; Liu, X.; Li, X.; Hu, Z.; Wang, L. Novel Harziane Diterpenes from Deep-Sea Sediment Fungus Trichoderma sp. SCSIOW21 and Their Potential Anti-Inflammatory Effects. Mar. Drugs 2021, 19, 689. [Google Scholar] [CrossRef]
- Wang, L.; Umezawa, K. Cellular Signal Transductions and Their Inhibitors Derived from Deep-Sea Organisms. Mar. Drugs 2021, 19, 205. [Google Scholar] [CrossRef]
- Wang, L.; Li, M.; Lin, Y.; Du, S.; Liu, Z.; Ju, J.; Suzuki, H.; Sawada, M.; Umezawa, K. Inhibition of cellular inflammatory mediator production and amelioration of learning deficit in flies by deep sea Aspergillus-derived cyclopenin. J. Antibiot. 2020, 73, 622–629. [Google Scholar] [CrossRef]
- Zou, J.-X.; Song, Y.-P.; Liu, X.-H.; Li, X.-N.; Ji, N.-Y. Bisabolane, cadinane, and cyclonerane sesquiterpenes from an algicolous strain of Trichoderma asperelloides. Bioorganic Chem. 2021, 115, 105223. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, S.-k.; Kang, J.S.; Choi, H.D.; Son, B.W. Polyketide and Sesquiterpenediol Metabolites from a Marine-Derived Fungus. Bull. Korean Chem. Soc. 2004, 25, 607–608. [Google Scholar] [CrossRef]
- Nozoe, S.; Goi, M.; Morisaki, N. Structure of cyclonerodiol. Tetrahedron Lett. 1970, 11, 1293–1296. [Google Scholar] [CrossRef]
- David, E.; Cane, R.I.; Shiao, M.-S. Cyclonerodiol Biosynthesis and the Enzymatic Conversion of Farnesyl to Nerolidyl pyrophosphate. J. Am. Chem. Soc. 1981, 103, 914–931. [Google Scholar]
- Guo, Y.W.; Gong, B.Q.; Yuan, J.; Li, H.J.; Mahmud, T.; Huang, Y.; Li, J.F.; Yang, D.P.; Lan, W.J. l-Phenylalanine Alters the Privileged Secondary Metabolite Production in the Marine-Derived Fungus Trichoderma erinaceum F1-1. J. Nat. Prod. 2020, 83, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yang, X.; Ji, Z.; Zhu, L.; Ma, N.; Chen, D.; Jia, X.; Tang, J.; Cao, Y. DFT-Calculated IR Spectrum Amide I, II, and III Band Contributions of N-Methylacetamide Fine Components. ACS Omega 2020, 5, 8572–8578. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.P.; Miao, F.P.; Liu, X.H.; Yin, X.L.; Ji, N.Y. Cyclonerane Derivatives from the Algicolous Endophytic Fungus Trichoderma asperellum A-YMD-9-2. Mar. Drugs 2019, 17, 252. [Google Scholar] [CrossRef]
- Song, Y.-P.; Miao, F.-P.; Liang, X.-R.; Yin, X.-L.; Ji, N.-Y. Harziane and cadinane terpenoids from the alga-endophytic fungus Trichoderma asperellum A-YMD-9-2. Phytochem. Lett. 2019, 32, 38–41. [Google Scholar] [CrossRef]
- Song, Y.-P.; Fang, S.-T.; Miao, F.-P.; Yin, X.-L.; Ji, N.-Y. Diterpenes and Sesquiterpenes from the Marine Algicolous Fungus Trichoderma harzianum X-5. J. Nat. Prod. 2018, 81, 2553–2559. [Google Scholar] [CrossRef]
- Adelin, E.; Servy, C.; Martin, M.-T.; Arcile, G.; Iorga, B.I.; Retailleau, P.; Bonfill, M.; Ouazzani, J. Bicyclic and tetracyclic diterpenes from a Trichoderma symbiont of Taxus baccata. Phytochemistry 2014, 97, 55–61. [Google Scholar] [CrossRef]
- Faylo, J.L.; van Eeuwen, T.; Kim, H.J.; Gorbea Colon, J.J.; Garcia, B.A.; Murakami, K.; Christianson, D.W. Structural insight on assembly-line catalysis in terpene biosynthesis. Nat. Commun. 2021, 12, 3487. [Google Scholar] [CrossRef]
- Hong, Y.J.; Tantillo, D.J. Consequences of conformational preorganization in sesquiterpene biosynthesis: Theoretical studies on the formation of the bisabolene, curcumene, acoradiene, zizaene, cedrene, duprezianene, and sesquithuriferol sesquiterpenes. J. Am. Chem. Soc. 2009, 131, 7999–8015. [Google Scholar] [CrossRef]
- Xu, Z.; Hong, L.L.; Liu, C.S.; Kong, J.Q. Protein Engineering of PhUGT, a Donor Promiscuous Glycosyltransferase, for the Improved Enzymatic Synthesis of Antioxidant Quercetin 3-O-N-Acetylgalactosamine. J. Agric. Food Chem. 2022, 70, 4076–4085. [Google Scholar] [CrossRef] [PubMed]
- Howard-Jones, A.R.; Kruger, R.G.; Lu, W.; Tao, J.; Leimkuhler, C.; Kahne, D.; Walsh, C.T. Kinetic analysis of teicoplanin glycosyltransferases and acyltransferase reveal ordered tailoring of aglycone scaffold to reconstitute mature teicoplanin. J. Am. Chem. Soc. 2007, 129, 10082–10083. [Google Scholar] [CrossRef] [PubMed]
- Ozenver, N.; Efferth, T. Small molecule inhibitors and stimulators of inducible nitric oxide synthase in cancer cells from natural origin (phytochemicals, marine compounds, antibiotics). Biochem. Pharmacol. 2020, 176, 113792. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).