Abstract
Five new fusarin derivatives, steckfusarins A–E (1–5), and two known natural products (6, 7), were isolated and identified from the marine algicolous fungus Penicillium steckii SCSIO 41040. The new compounds, including absolute configurations, were determined by spectroscopic analyses and calculated electronic circular dichroism (ECD). All new compounds were evaluated for their antioxidant, antibacterial, antifungal, antiviral, cytotoxic, anti-inflammatory, antioxidant, cholesterol-lowering, acetyl cholinesterase (AChE) enzyme and 6-phosphofructo-2-kinase (PFKFB3) and phosphatidylinositol-3-kinase (PI3K) inhibitory activities. The biological evaluation results revealed that compound 1 exhibited radical scavenging activity against 2,2-diphenyl-1-picrylhydrazylhydrate (DPPH), with an IC50 value of 74.5 µg/mL. In addition, compound 1 also showed weak anti-inflammatory activity at a concentration of 20 µM.
1. Introduction
Based on genome sequencing and mining and antimicrobial screening of crude extracts, P. steckii has shown strong activity against some pathogenic bacteria [1]. It has been recently reported that the secondary metabolites of P. steckii were mainly tanzawaic acids and alkaloids that demonstrated a broad range of significant biological activities, such as antimicrobial, anti-inflammatory and lipid-lowering activities [2,3,4,5,6].
Fusarins are a class of mycotoxins characterized by a 2-pyrrolidone moiety and a triene side chain of twelve carbon atoms [7] that are usually isolated from Penicillium sp. [8]. The compounds were biosynthesized by a hybrid polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) [9]. In the human neuroblastoma cell line SH-SY5Y, fusarin derivatives were first proven to effectively promote neurite growth and block the cell cycle in the G0/G1 phase [10]. Fusarin derivatives inhibit mammalian DNA polymerases and human DNA topoisomerase II in vitro [11]. Additionally, they induce the formation of 6-thioguanine-resistant mutants in V79 cells, as well as the asynchronous replication of polyoma DNA sequences subsequent to the presence of a microsomal activation system [12,13]. Fusarins have been reported to be unstable after prolonged exposure to ultraviolet light and elevated temperatures [14]; additionally, light can induce the rearrangement of fusarins, leading to the simultaneous appearance of fusarin analogues [15].
During our continuous study exploring new bioactive natural products from marine microorganisms [16,17], five new fusarin derivatives (1–5) (Figures S1–S4) and two known natural products (6, 7) (Figure 1) were obtained from the fungus Penicillium steckii SCSIO 41040, which was isolated from a green algae Botryocladia sp. of the South China Sea. Herein, we describe the fermentation, isolation, structural determination and biological activities of these compounds.
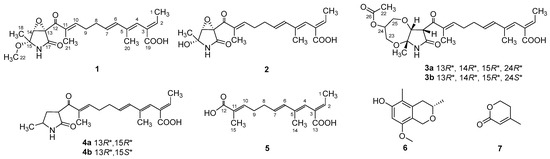
Figure 1.
Chemical structures of compounds 1–7.
2. Results and Discussion
Compound 1 was obtained as a yellow oil, and the molecular formula C21H27NO6 was assigned to it based on its HRESIMS (m/z 390.1912 [M + H]+, calcd for C21H28NO6+, 389.1911), which required nine degrees of unsaturation. The 1H NMR and 13C NMR data (Table 1) indicated five olefinic methines (δC/H 138.5/6.80, CH-2; 123.5/5.94, CH-4; 134.9/6.25, CH-6; 128.4/5.73, CH-7; 148.0/6.86, CH-10), five methyls (δC/H 15.6/1.66, CH3-1; 18.7/1.39, CH3-18; 14.2/1.56, CH3-20; 10.8/1.75, CH3-21; 49.5/3.26, CH3-22), two methylenes (δC/H 31.0/2.26, CH2-8; 28.8/2.42, CH2-9), one oxygenated methine (δC/H 62.4/4.28, CH-14) and eight quaternary carbonyls (δC 131.0, C-3; 136.7, C-5; 137.5, C-11; 190.0, C-12; 62.2, C-13; 87.4, C-15; 169.9, C-17; 167.9, C-19). Seven degrees of unsaturation could be accounted for by the presence of three carbonyl signals (a ketone resonance (δC 190.0), an amide (δC 169.9) and a carboxyl (δC 167.9)) and the eight sp2 carbon signals of four olefinic bonds. Therefore, the remaining unsaturation indicated the presence of two rings. HMBC correlations from H-14 to C-15 and NH-16 to C-13/C-14/C-15/C-17 established the presence of the 2-pyrrolidone ring structure. The highly deshielded chemical shift of C-13 and CH-14 inferred that both were oxygenated, which prompted us to speculate on the existence of an epoxide ring in the 2-pyrrolidone ring. Additionally, the location of the methyl and methoxyl groups were determined via HMBC correlations from H3-18 to C-14/C-15 and H3-22 to C-15. According to the 2D NMR spectra, a 2-ethylidene-4,10-dimethyl-11-oxoundeca-3,5,9-trienoic acid side chain was located at C-13 of the 2-pyrrolidone ring, supported by COSY correlations of H3-1/H-2 and H-6/H-7/H2-8/H2-9/H-10 and HMBC correlations from H3-1 to C-3, H-2 to C-19, H-4 to C-2/C-6, H3-20 to C-4/C-5 and H3-21 to C-10/C-11/C-12. The E-configuration of the ∆6(7) double bond was determined by the coupling constant (J = 15.6 Hz). NOESY correlations indicated an E-configuration for the other double bonds in the side chains of H3-1/H-4, H-4/H-6, H2-9/H3-21 and the cofacial orientation of H3-18/H-14. The presence of an epoxide ring indicated the homoconfiguration of H-13/H-14. The absolute configurations of compound 1 were determined by ECD calculations. The experimental ECD spectra were compared with two calculated spectra, (13R, 14R, 15R)-1 and (13S, 14R, 15R)-1. The results showed that the experimental ECD spectra of 1 matched well with the calculated spectrum for (13R, 14R, 15R)-1, confirming the absolute configurations of 1 as depicted (Figure 2).

Table 1.
1H and 13C NMR spectroscopic data for compounds 1–3 and 5 (700, 175 MHz, DMSO-d6).
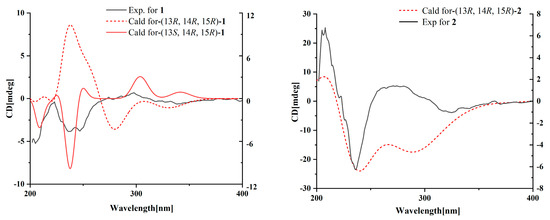
Figure 2.
Experimental and calculated ECD spectra for compounds 1 and 2.
Compound 2 was isolated as a yellow oil. The HRESIMS (m/z 347.1609 [M − H]−, calcd for C20H24NO6−, 374.1609) data suggested a molecular formula of C20H25NO6, revealing nine degrees of unsaturation. The NMR signals (Table 1) of 2 resembled those of 1, except for the absence of a methoxyl group at δC/H 49.5/3.26. Large coupling (J = 15.6 Hz) between H-6 (δH 6.25) and H-7 (δH 5.74) showed that the ∆6(7) olefinic bond was in E-configuration. NOESY data of H3-1 (δH 1.65)/H-4 (δH 5.93), H-4 /H-6 and H2-9 (δH 2.40)/H3-21 (δH 1.75), indicated that the ∆2(3), ∆4(5) and ∆10(11) olefinic bonds were all in E-configuration. The NOESY spectrum of 2 showed key correlations of H3-18/H-14, indicating that they were co-facial. The high similarity between the calculated ECD curve of (13R, 14R, 15R)-2 and its experimental curve unambiguously confirmed the absolute configuration of 2 (Figure 2) [18,19,20].
Compound 3 was obtained as a pink oil. Its molecular formula of C25H33NO8 was determined via HRESIMS (m/z 476.2290 [M + H]+, calcd for C25H34NO8+, 476.2279), which required ten degrees of unsaturation. Proton network systems of H-13 (δH 4.93)/H-14 (δH 4.33) and H2-23 (δH 3.51, 3.67)/H-24 (δH 4.16)/H2-25 (δH 3.34, 3.99) were detected in the COSY spectrum. Moreover, HMBC correlations from H-13 to C-17 (δC 170.5), NH-16 (δH 8.89) to C-13 (δC 48.8)/C-14 (δC 77.2)/C-15 (δC 82.9)/C-17, H3-18 (δH 1.48) to C-14/C-15 and H2-23 to C-15 established the presence of a glycerol moiety and the 2-pyrrolidone ring structure, which were connected via ether linkages to form a 8a-methyl-2-oxohexahydro-1H,5H-[1,4]dioxepino [2,3-b]pyrrol-6-yl acetate ring system (Figure 3). A detailed analysis of its NMR spectroscopic features implied that it was closely structurally related to dothilactaene B [15]. A set of signals at CH3-22 and C-26 (Table 1 and Table 2), which were not present in the 1H and 13C NMR spectra of dothilactaene B, were observed in the spectrum of 3. Meanwhile, compound 3 lacked a methoxy group at δC/H 51.9/3.55. Through the analyses of the HMBC spectrum, the acetic acid group was confirmed by the HMBC correlations from H3-22 (δH 1.98) to C-26 (δC 170.8) and H2-25 to C-26. The E-configuration of the ∆ 6(7) double bond was determined by the coupling constant (J = 15.6 Hz). NMR data analyses of 3 showed a similar multiplicity pattern in the glycerol moiety, indicating that they are two components and have the same plane structure. The resonances of the glycerol moiety suggested them to be diastereoisomers presenting in a ratio of 3:1 (3a:3b), which may be formed by Michael addition [21]. NOESY correlations (Figure 4) of 3a indicated the E-configuration for the other double bonds in the side chain and the cofacial orientation of H-13, H-14, CH3-18 and H-24. The NOESY spectrum of 3b was the same as 3a, except the signal of H-24 was the opposite. These data permitted assignment of the (13R*, 14R*, 15R*, 24R*)-3a and (13R*, 14R*, 15R*, 24S*)-3b relative configurations [15,19].
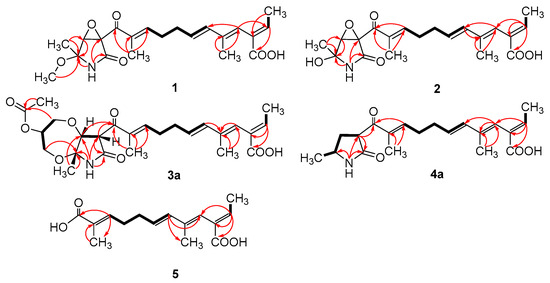
Figure 3.
Key COSY (bold lines) and HMBC (arrows) correlations of compounds 1–5.
Table 2.
Partial 1H and 13C NMR spectroscopic data for 3 (700, 175 MHz, DMSO-d6).
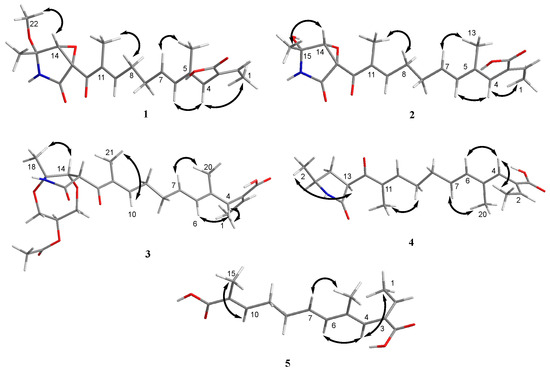
Figure 4.
Key NOESY (double arrow) correlations of compounds 1–5.
Compound 4 was obtained as a yellow oil. Its HRESIMS ([M + H]+, 346.2016; calcd for C20H28NO4+, 346.2013) data were in agreement with the molecular formula C20H27NO4. Its 1H NMR and 13C NMR data (Table 3) closely resembled the data for 2, except for the absence of the hydroxyl group and an epoxy system at the 2-pyrrolidone ring structure. To data, all fusarin derivatives reported from natural sources have had a free hydroxyl group at C-15; however, compound 4 has no such substituent [22,23,24]. The relative configuration of 4 was confirmed by its NOESY correlations and coupling constants (Figure 4). The NOESY correlations of H3-1 (δH 1.64)/H-4 (δH 5.95), H-4/H-6 (δH 6.26), H2-9 (δH 2.38)/H3-21 (δH 1.72) and the large coupling constant of H-6/H-7 (J = 15.6 Hz) suggested the E geometry of all double bonds. Each pair of 1H and 13C NMR signals showed a similar multiplicity pattern, suggesting that compound 4 was an inseparable mixture of two geometric isomers presenting in a ratio of 1:1 (4a:4b). Even under mild conditions, the stereo center of C-15 was easy to be epimerized through ring opening [19,20]. Comparing 4a and 4b (Table 2), the main difference occurred at the configuration at C-13, C-15 and C-18, which was proven by the carbon chemical shifts of C-13 (∆δC 1.1 ppm), C-15 (∆δC 1.1 ppm) and C-18 (∆δC 0.2 ppm). The NOESY spectrum of 4a showed cross-peaks between H-13 (δH 4.33) and H3-18 (δH 1.11) that indicated H-13 and H-15 (δH 1.48) were on the opposite side, whereas no relevant signal between H-13 and H3-18 was found in 4b. Accordingly, the relative configurations of 4a and 4b could be assigned as (13R*, 15R*)-4a and (13R*, 15S*)-4b.
Table 3.
1H and 13C NMR spectroscopic data for 4 (700, 175 MHz, DMSO-d6).
Compound 5 was obtained as a yellow oil. It showed a [M + H]+ ion peak at m/z 265.1431 in the positive-ion HRESIMS (calcd. for C15H21O4+, 265.1434) that was appropriate for a molecular formula of C15H20O4. The NMR data of 5 showed that it shared the same trienedioic acid skeleton as epolactaene 4a [25]. The main difference was the absence of a methyl in 5. The large vicinal coupling constant (J = 15.6 Hz) between H-6 (δH 6.23) and H-7 (δH 5.72) suggested an E-configuration for the ∆ 6(7) double bond. In addition, the NOESY correlations between H3-1 (δH 1.65) and H-4 (δH 5.94), H-4 and H-6 and H2-9 (δH 2.45) and H3-15 (δH 1.74) indicated an E-configuration for the other double bonds (Figure 4). Therefore, the structure of 5, named as steckfusarin F, was elucidated.
The other two known compounds were elucidated as (3S)-3,5-dimethyl-8-methoxy-3,4-dihydro-1H-isochromen-6-ol (6) [26] and 4-methyl-5,6-dihydro-2H-pyran-2-one (7) [27] by comparing their NMR and MS data with the data reported in the literature.
The new compounds (1–5) were evaluated for antioxidant, antibacterial, antifungal, antiviral, cytotoxic, anti-inflammatory, cholesterol-lowering and PFKFB3 and PI3K kinase inhibitory activities in vitro.
The isolated compounds (1–5) were evaluated for their cytotoxic activities against twenty human cancer cell lines (A549, MKN-45, HCT 116, HeLa, K-562, MCF7, HepG2, SF126, DU145, CAL-62, 786-O, TE-1, 5637, GBC-SD, L-02, PATU8988T, HOS, A-375, A-673 and 293T) via CCK-8 (Dojindo) method. However, none of the compounds showed obvious cytotoxicity. Likewise, none of the new compounds did exhibited any growth inhibition when tested against Klebsiella pneumonia, Candida albicans Berkhout, Staphyloccocus aureus, Colletotrichum gloeosporioiles, Magnaporthe grisea and Clerotinia miyabeana Hanzawa. In addition, none of the new compounds exhibited cholesterol-lowering or PFKFB3 and PI3K kinase inhibitory activities. Compound 1 showed weak inhibitory activity against lipopolysaccharide-inducted nitric oxide (NO) in RAW 264.7 cells at a concentration of 20 µM.
The radical scavenging activities of all the isolated compounds were tested against DPPH; compounds 1, 2 and 4 showed radical scavenging activity against DPPH, with 37.3%, 54.3% and 45.8% inhibition at concentrations of 100 µg/mL, respectively. Due to the low mass of the active compounds, only compound 1 was measured for IC50. It had an IC50 value of 74.5 µg/mL, whereas ascorbic acid, used as a positive control, had an IC50 value of 7.5 µg/mL. Furthermore, molecular docking analyses between the active compounds and superoxide dismutase (PDB ID: 7wx0) were performed to gain functional and structural insights (Figure 5). The results showed that compound 1 could interact with superoxide dismutase at the entrance of the catalytic pocket, with a calculated binding affinity of −6.3 kcal/mol. Compound 1 interacted with the residues SER-32 and GLN-153 via two hydrogen bonds, and five hydrophobic interactions were formed between compound 1 and the residues LYS-4, VAL-6, HIS-20, ALA-152 and GLN-153. The active site of 1 contained methoxy and carboxyl groups. The molecular docking results demonstrated that the negative binding free energy value between compound 2 and superoxide dismutase was −6.6 kcal/mol. Interaction modes revealed six hydrogen bonding interactions with the residues GLN-23, ARG-79, ARG-79, SER-102, LEU-103 and ILE-104. In addition, there were two hydrophobic interactions with LEU-103 and ALA-105 in the active site of superoxide dismutase. The active site of 2 contained hydroxyl and carboxyl groups and an epoxy ring. The size of compound 1 was 51 × 51 × 47, centered at x: 17.894, y: 7.9, z: 82.091. The size of compound 2 was 49 × 44 × 49, centered at x: 17.894, y: 7.9, z: 82.091. Compounds 1 and 2 had no π-π stacking and π-cation interactions. The docking studies suggested that compounds 1 and 2 could inhibit superoxide dismutase by tightly binding to catalytic amino acid residues through different types of interactions.
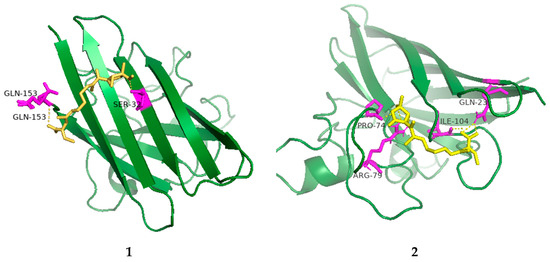
Figure 5.
Low-energy binding conformations of 1 and 2 bound to superoxide dismutase (generated by molecular docking).
Although many activities of fusarin derivatives have been reported [4,8,13,17], no obvious activities were detected for compounds 1–5. It was reported that the tetrahydropyran ring and methylation of the acid group from the sidechain were crucial to the activity of these compounds [21]. However, none of the new compounds were methylated at C-19, which may be the reason why they did not display obvious activities.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were recorded using a Perkine Elmer 341 polarimeter (Hertford, UK). ECD spectra were recorded with a Chirascan circular dichroism spectrometer (Applied Photophysics, Surrey, UK). UV spectra were recorded on a UV-2600 UV–vis spectrophotometer (Shimadzu, Kyoto, Japan). The 1D and 2D NMR spectra were recorded on a Bruker AC 500 and 700 NMR (Broker, Fallanden, Switzerland) spectrometer with TMS as the internal standard. HRESIMS spectra were measured with a Bruker micro TOF-QII (Bruker, Fallanden, Switzerland) mass spectrometer in positive/negative ion mode. Silica gel GF-254 (10–40 mm) was used for thin-layer chromatography (TLC) (Qingdao Marine Chemical Factory, Qingdao, China). Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) and silica gel (200–300 mesh, 100–200 mesh) (Qingdao Marine Chemical Factory, Qingdao, China) were applied in column chromatography (CC). HPLC was carried out on a Hitachi Primaide with a YMC ODS Series column (YMC-Pack ODS-A, YMC Co. Ltd. (Kyoto, Japan), 250 × 10 mm i.d., S-5 μm, 12 nm). All solvents were analytical grade (Tianjin Fuyu Chemical and Industry Factory). The fermentation culture medium and reagents were obtained from Guangzhou Haili Aquarium Technology Company, Guangzhou, China.
3.2. Fungal Strain
The fungal strain Penicillium steckii SCSIO 41040 was isolated from a green algae Botryocladia sp. that was collected from the South China Sea. The isolated fungal strain was stored on Muller Hinton broth (MB) agar (malt extract 16 g, agar 18 g, sea-salt 30 g, water 1 L and pH 7.4–7.8) slants at 4 °C and then deposited in CAS Key Laboratory of Tropical Marine Bio-resources and Ecology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, PR China. The ITS1-5.8S-ITS2 sequence region (550 base pairs (bp), GenBank accession no. OP349656) of strain SCSIO 41040 was amplified via the PCR process. DNA sequencing showed that it shared significant homology (100%) with Penicillium steckii (GenBank accession no. NR111488).
3.3. Fermentation and Extraction
A few loops of cells of the strain SCSIO 41040 were inoculated into a 500 mL Erlenmeyer flask containing 150 mL of seed medium (malt extract 1%, yeast extract 0.4%, glucose 0.4% and pH 7.2) and cultivated on a rotary shaker at 180 rpm and 28 °C for 48 h as a seed culture. Then, a large-scale fermentation of the fungal strain SCSIO 41040 was incubated at 25 °C in 1 L conical flasks containing a solid medium (300 mL/flask) composed of 200 g rice and 220 mL 3.2% (NaCl 3.2 g/H2O 100 mL) artificial seawater. After 32 days, the fermented material from each flask was extracted successively with EtOAc (700 mL/flask) and the combined EtOAc extract was suspended in MeOH and extracted with petroleum ether to remove rice oil. Finally, the MeOH solution was concentrated under reduced pressure to obtain a reddish-brown extract (56.0 g).
3.4. Isolation and Purification
The extract was subjected to silica gel column chromatography (CC), eluting with a gradient CH2Cl2-MeOH (100:0–0:100, v/v) to give 15 fractions based on TLC properties. Fr.3 was separated by semipreparative HPLC (40% MeCN/H2O + 0.3% TFA, 2.0 mL/min) to provide 6 (10.7 mg, tR 17 min) and 7 (9.2 mg, tR 8 min). Fr.5 was separated into 8 subfractions (Fr.5.1–5.8) using ODS silica gel chromatography via elution with MeOH/H2O (5–100%). Fr.5.6 was directly separated via semipreparative HPLC (56% MeOH/H2O + 0.3% TFA, 2.7 mL/min) to offer 4 (4.5 mg, tR 44 min). Fr.5.7 was separated by semipreparative HPLC (35% MeOH/H2O + 0.3% TFA, 2.7 mL/min) to give 5 (8.5 mg, tR 37 min). Fr.6 was applied to ODS silica gel chromatography to give eight fractions (Fr.6.1–6.8). Fr.6.2 and Fr.6.3 were further divided into 6 (Fr.6.2.1–Fr.6.2.6) and 11 (Fr.6.3.1–Fr.6.3.11) parts through a Sephadex LH-20 with MeOH and semipreparative HPLC (56% MeOH/H2O + 0.3% TFA, 2.5 mL/min), respectively. Fr.6.2.2 was purified by semipreparative HPLC (58% MeOH/H2O + 0.3% TFA, 2.7 mL/min) to yield 3 (3.0 mg, tR 15 min). Fr.6.2.6 was separated by semipreparative HPLC (37% MeCN/H2O + 0.3% TFA, 3.0 mL/min) to yield 1 (23.2 mg, tR 42 min). Fr.6.3.10 was directly separated via semipreparative HPLC (34% MeCN/H2O + 0.3% TFA, 2.5 mL/min) to provide 2 (5.8 mg, tR 33 min).
Steckfusarin A (1): yellow oil; − 14.8°; UV (MeOH) λmax (log ε) 235 (3.94) nm; 1H and 13C NMR data, Table 1; HRESIMS m/z 390.1912 [M + H]+ (calcd for C21H28NO6+, 389.1911).
Steckfusarin B (2): yellow oil; + 24.4°; UV (MeOH) λmax (log ε) 213 (4.27), 233 (4.27) nm; 1H and 13C NMR data, Table 1; HRESIMS m/z 347.1609 [M − H]− (calcd for C20H24NO6−, 374.1609).
Steckfusarin C (3): pink oil; − 1.7°; UV (MeOH) λmax (log ε) 212 (4.03), 232 (4.01) nm; 1H and 13C NMR data, Table 1 and Table 2; HRESIMS m/z 476.2290 [M + H]+ (calcd for C25H34NO8+, 476.2279).
Steckfusarin D (4): yellow oil; – 8.5°; UV (MeOH) λmax (log ε) 204 (4.21) nm; 1H and 13C NMR data, Table 2; HRESIMS m/z 346.2016 [M + H]+ (calcd for C20H28NO4+, 346.2013).
Steckfusarin E (5): yellow oil; UV (MeOH) λmax (log ε) 216 (4.14) nm; 1H and 13C NMR data, Table 1; HRESIMS m/z 265.1431 [M + H]+ (calcd for C15H21O4+, 265.1434).
3.5. ECD Calculation
The relative configurations of the new compounds were subjected to random conformational searches using Spartan’14 and Gaussian 09 software with the Merck molecular force field (MMFF) and density functional theory (DFT)/TDDFT, respectively. The MMFF conformational search produced low-energy conformers with a Boltzmann population of more than 5%, which were geometrically optimized using the DFT method at the B3LYP/6-311G* level in MeOH using the IEFPCM model. For the stable conformers of the new compounds, the overall theoretical calculation of ECD was achieved in MeOH using time-dependent density functional theory at the B3LYP/6-311G* level. We used the 0.2–0.4 eV half bandwidth Multiwfn to generate the ECD spectra of different conformations and calculated the contribution according to the Boltzmann of each conformation after the UV correction [28].
3.6. Cell Culture and Cytotoxic Bioassay
The obtained fusarin analogues (1–5) were evaluated for their cytotoxic activities against twenty cell lines, A549, MKN-45, HCT 116, HeLa, K-562, MCF7, HepG2, SF126, DU145, CAL-62, 786-O, TE-1, 5637, GBC-SD, L-02, PATU8988T, HOS, A-375, A-673 and 293T (Shanghai Cell Bank, Chinese Academy of Sciences). The cytotoxic activity was determined via the CCK-8 (Dojindo) method [29].
3.7. Antimicrobial Assay
All new compounds were tested for antibacterial activity against Klebsiella pneumonia, Candida albicans Berkhout, Staphyloccocus aureus, Colletotrichum gloeosporioiles, Magnaporthe grisea and Clerotinia miyabeana Hanzawa. The tests were performed in 96-well plates using a modification of the broth microdilution method [30].
3.8. Anti-Inflammatory Assay
All new compounds were evaluated for their inhibitory activity against LPS-induced NF-κB activation in RAW264.7 cells using a luciferase reporter gene assay [31]. The RAW264.7 cells, stably transfected with the NF-κB luciferase reporter gene, were plated in triplicate into 96-well plates for all treatments and controls. The compounds (20 µM) and BAY11-7082 (an NF-κB inhibitor used as a positive control (5 µM); Sigma-Aldrich, St. Louis, MO, USA) were used to pretreat the cells for 30 min, followed by stimulation with 5 µg/mL LPS for 8 h. The cells were harvested and luciferase activity measured using a luciferase assay system (Promega, Madison, WI, USA).
3.9. Cholesterol Transport Mechanism
Pancreatic triglyceride lipase (PTL) and Niemann-Pick C1-like 1 (NPC1L1) were the crucial targets involved in cholesterol cellar uptake. The inhibitory activity of the compounds against PTL was evaluated by colorimetry. Surface plasmon resonance (SPR) was used to analyze the binding of the compounds with NPC1L1, and the activities of targeted compounds for NPC1L1 were studied [32,33].
3.10. PFKFB3 Kinase Inhibitory Activity
PFKFB3 kinase inhibitory activity was measured using an ADP-Glo Kinase Assay kit (Promega) according to a published modified protocol [34]. Compound solution (1 μL, with a final concentration of 20 μM in 1% DMSO) and 2 μL enzyme solution were added to 384-well plates, followed by incubation at room temperature (RT) for 30 min. After 2 h incubation at RT, 4 μL ADP-Glo Reagent was added and then the incubation continued for 1 h prior to the addition of 8 μL Kinase Detection Reagent and a further incubation of 1 h. The luminous signal was measured using an Envision flat panel reader (PerkinElmer, Waltham, MA, USA).
3.11. PI3K Kinase Inhibitory Activity
An amount of 0.5 μL of the compounds was preincubated with 14.5 μL of enzyme and PIP2 substrate for 10 min before addition of 5 μL of ATP to achieve a final ATP concentration of 10 μM [35]. The total reaction volume was 20 μL, and the reaction was allowed to proceed for 45 min at room temperature before the addition of the stop solution and detection mixture provided in the kit. The luminous signal was measured using an Envision flat panel reader (PerkinElmer).
3.12. Measurement of AChE Inhibition Activity
We evaluated the AChE enzyme inhibitory activity of compounds according to the slightly modified spectrophotometric method [36]. Tacrine was used as a positive control.
3.13. Molecular Docking Analysis
The crystal structure of neuraminidase (PDB ID: 7wx0) was retrieved from the Protein DataBank. We used AutoDockTools (Version 1.5.7) to conduct the molecular docking. The structures were generated in ChemBio3D Ultra 14.0 (ChemBioOffice version 14.0), followed by an MM2 calculation to minimize the conformation energy. The docking pose that had the lowest binding energy was represented as the most favorable binding conformation.
4. Conclusions
In this study, five new fusarin derivatives were obtained from the marine algicolous fungus Penicillium steckii SCSIO 41040, which had been fermented using rice solid medium. The new structures, including absolute configurations, were determined using spectroscopic methods coupled with the calculated ECD. Bioassays were used to screen the antioxidant, antibacterial, antifungal, antiviral, cytotoxic, anti-inflammatory, cholesterol-lowering and PFKFB3 and PI3K kinase inhibitory activities. Compound 1 exhibited antioxidant activity against DPPH, with an IC50 value of 74.5 µg/mL. In addition, compound 1 showed weak anti-inflammatory activity at a concentration of 20 µM. Compared with the previously reported fusarin derivatives [10,14,19,23], compounds 1–5 have no obvious antitumor activities, possibly because the new compounds lacked methylation at C-19.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/md21100532/s1. Figures S1–S47: 1H, 13C, DEPT135, COSY, HSQC, HMBC, NOESY, HRESIMS and UV spectra of the new compounds 1–5. Tables S1 and S2: Energies of 1 and 2 at MMFF94 force field and B97-3c level.
Author Contributions
Y.S., J.S. and W.C.; formal analysis, J.W. (Jiamin Wang) and Y.S.; investigation, J.S. and X.Z.; project administration, Y.T. and X.P.; resources, J.W. (Junfeng Wang) and Y.L.; writing original draft, Y.S.; writing-review and editing, J.W. (Junfeng Wang). All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Guangdong Basic and Applied Basic Research Foundation (2021B1515120046, and 2021A1515011523), Guangdong Local Innovation Team Program (2019BT02Y262), the Hainan Provincial Joint Project of Sanya Yazhou Bay Science and Technology City (Grant Nos. 2021JJLH0097 and 2021CXLH0013) and Hainan Provincial Natural Science Foundation of China (823CXTD393).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to ZH Xiao, AJ Sun, XH Zheng, Y Zhang and X Ma in the analytical facility at SCSIO for recording the spectroscopic data and to WZ Xiang for providing the samples and pictures of the green algae Botryocladia sp.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yao, G.; Chen, X.; Zheng, H.; Liao, D.; Yu, Z.; Wang, Z.; Chen, J. Genomic and chemical investigation of bioactive secondary metabolites from a marine-derived fungus Penicillium steckii P2648. Front. Microbiol. 2021, 12, 600911. [Google Scholar] [CrossRef]
- Hu, X.; Li, X.; Wang, B.-G.; Meng, L.-H. Uncommonp polyketides from Penicillium steckii AS-324, a marine endozoic fungus isolated from deep-sea coral in the magellan seamount. Int. J. Mol. Sci. 2022, 23, 6332. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, X.; Wang, B.; Meng, L. Tanzawaic acid derivatives: Fungal polyketides from the deep-sea coral-derived endozoic Penicillium steckii AS-324. J. Nat. Prod. 2022, 85, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, W.; Pang, X.; Liao, S.; Wang, J.; Lin, X.; Yang, B.; Zhou, X.; Luo, X.; Liu, Y. Pyrrolyl 4-quinolone alkaloids from the mangrove endophytic fungus Penicillium steckii SCSIO 41025: Chiral resolution, configurational assignment, and enzyme inhibitory activities. Phytochemistry 2021, 186, 112730. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tan, Y.; She, J.; Chen, C.; Wang, J.; Hu, Y.; Pang, X.; Wang, J.; Liu, Y. Tanzawaic acid derivatives from the marine-derived Penicillium steckii as inhibitors of RANKL-Induced osteoclastogenesis. J. Nat. Prod. 2023, 86, 1171–1178. [Google Scholar] [CrossRef]
- Shin, H.; Pil, G.; Heo, S.-J.; Lee, H.-S.; Lee, J.; Lee, Y.-J.; Lee, J.; Won, H. Anti-inflammatory activity of tanzawaic acid derivatives from a marine-derived fungus Penicillium steckii 108YD142. Mar. Drugs 2016, 14, 14. [Google Scholar] [CrossRef]
- Maragos, C.M.; Busman, M.; Plattner, R.D. Development of monoclonal antibodies for the fusarin mycotoxins. Food Addit. Contam. 2008, 25, 105–116. [Google Scholar] [CrossRef]
- Nagumo, Y.; Kakeya, H.; SHOJI, M.; Hayashi, Y.; Dohmae, N.; Osada, H. Epolactaene binds human Hsp60 Cys442 resulting in the inhibition of chaperone activity. Biochem. J. 2005, 387, 835–840. [Google Scholar] [CrossRef]
- Donzelli, B.G.; Krasnoff, S.B.; Churchill, A.C.; Vandenberg, J.D.; Gibson, D.M. Identification of a hybrid PKS-NRPS required for the biosynthesis of NG-391 in Metarhizium robertsii. Curr. Genet. 2010, 56, 151–162. [Google Scholar] [CrossRef]
- Kakeya, H.; Onozawa, C.; Sato, M.; Arai, K.; Osada, H. Neuritogenic effect of epolactaene derivatives on human neuroblastoma cells which lack high-affinity nerve growth factor receptors. J. Med. Chem. 1997, 40, 391–394. [Google Scholar] [CrossRef]
- Kuramochi, K.; Yukizawa, S.; Ikeda, S.; Sunoki, T.; Arai, S.; Matsui, R.; Morita, A.; Mizushina, Y.; Sakaguchi, K.; Sugawara, F.; et al. Syntheses and applications of fluorescent and biotinylated epolactaene derivatives: Epolactaene and its derivative induce disulfide formation. Bioorg. Med. Chem. 2008, 16, 5039–5049. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.J.; Ronai, Z.A.; Li, M.H.; Jeffrey, A.M. Fusarium moniliforme metabolites: Genotoxicity of culture extracts. Carcinogenesis 1988, 9, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.J.; Jiang, Y.Z.; Li, M.H.; Lo, H.Z.A. A mutagenic metabolite produced by Fusarium moniliforme isolated from Linxian county, China. Carcinogenesis 1985, 6, 903–905. [Google Scholar] [CrossRef]
- Sondergaard, T.E.; Hansen, F.T.; Purup, S.; Nielsen, A.K.; Bonefeld-Jorgensen, E.C.; Giese, H.; Sorensen, J.L. Fusarin C acts like an estrogenic agonist and stimulates breast cancer cells in vitro. Toxicol. Lett. 2011, 205, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Kleigrewe, K.; Aydin, F.; Hogrefe, K.; Piecuch, P.; Bergander, K.; Wurthwein, E.U.; Humpf, H.U. Structure elucidation of new fusarins revealing insights in the rearrangement mechanisms of the Fusarium mycotoxin fusarin C. J. Agric. Food Chem. 2012, 60, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Su, Z.; Gao, Y.; Lin, X.; Pang, X.; Yang, B.; Tao, H.; Luo, X.; Liu, Y.; Zhou, X. Cytotoxic minor piericidin derivatives from the actinomycete strain Streptomyces psammoticus SCSIO NS126. Mar. Drugs. 2021, 19, 428. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Lin, X.; Salendra, L.; Pang, X.; Dai, Y.; Yang, B.; Liu, J.; Wang, J.; Zhou, X.; Liu, Y. Isobenzofuranones and isochromenones from the deep-sea derived fungus Leptosphaeria sp. SCSIO 41005. Mar. Drugs 2017, 15, 204. [Google Scholar] [CrossRef]
- Kakeya, H.; Takahashi, I.; Okada, G.; Isono, K.; Osada, H. Eolactaene, a novel neuritogenic compound in human neuroblastoma cells, produced by a marine fungus. J. Antibiot. 1995, 48, 733–735. [Google Scholar] [CrossRef]
- Kakeya, H.; Kageyama, S.; Nie, L.; Onose, R.; Okada, G.; Beppu, T.; Norbury, C.J.; Osada, H. Lucilactaene, a new cell cycle inhibitor in p53-transfected cancer cells, produced by a Fusarium sp. J. Antibiot. 2011, 54, 850–854. [Google Scholar] [CrossRef][Green Version]
- Yamaguchi, J.; Kakeya, H.; Uno, T.; Shoji, M.; Osada, H.; Hayashi, Y. Determination by asymmetric total synthesis of the absolute configuration of lucilactaene, a cell-cycle inhibitor in p53-transfected cancer cells. Angew. Chem. Int. Ed. Engl. 2005, 44, 3110–3115. [Google Scholar] [CrossRef]
- Kumarihamy, M.; Rosa, L.H.; Duke, S.O.; Tekwani, B.L.; Techen, N.; Ferreira, D.; Croom, E.M.; Khan, S.; Nanayakkara, N.P.D. Antimalarials and phytotoxins from Botryosphaeria dothidea identified from a seed of diseased Torreya taxifolia. Molecules 2021, 26, 59. [Google Scholar] [CrossRef] [PubMed]
- Kleigrewe, K.; Sohnel, A.C.; Humpf, H.U. A new high-performance liquid chromatography-tandem mass spectrometry method based on dispersive solid phase extraction for the determination of the mycotoxin fusarin C in corn ears and processed corn samples. J. Agric. Food. Chem. 2011, 59, 10470–10476. [Google Scholar] [CrossRef] [PubMed]
- Abdelhakim, I.A.; Mahmud, F.B.; Motoyama, T.; Futamura, Y.; Takahashi, S.; Osada, H. Dihydrolucilactaene, a potent antimalarial compound from Fusarium sp. RK97-94. J. Nat. Prod. 2021, 85, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, W.C.A.; Marasas, W.F.O.; Steyn, P.S.; Thiel, P.G.; Merwe, K.J.v.d.; Rooyen, P.H.v.; Vleggaar, R.; Wesselsb, P.L. Structure Eelucidation of fusarin C, a mutagen produced by Fusarium moniliforme. J. Chem. Soc. Chem. Commun. 1984, 2, 122–124. [Google Scholar]
- Kuramochi, K.; Sunoki, T.; Tsubaki, K.; Mizushina, Y.; Sakaguchi, K.; Sugawara, F.; Ikekita, M.; Kobayashi, S. Transformation of thiols to disulfides by epolactaene and its derivatives. Bioorg. Med. Chem. 2011, 19, 4162–4172. [Google Scholar] [CrossRef]
- Smetanina, O.F.; Yurchenko, A.N.; Pivkin, M.V.; Yurchenko, E.A.; Afiyatullo, S.S. Isochromene metabolite from the facultative marine fungus Penicillium citrinum. Chem. Nat. Compd. 2011, 47, 118–119. [Google Scholar] [CrossRef]
- Ying, Y.; Zhang, L.; Shan, W.; Zhan, J. Secondary metabolites of Peyronellaea sp. XW-12, an endophytic fungus of Huperzia serrata. Chem. Nat. Compd. 2014, 50, 723–725. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Tan, Y.; Yang, B.; Lin, X.; Luo, X.; Pang, X.; Tang, L.; Liu, Y.; Li, X.; Zhou, X. Nitrobenzoyl sesquiterpenoids with cytotoxic activities from a marine-derived Aspergillus ochraceus fungus. J. Nat. Prod. 2018, 81, 92–97. [Google Scholar] [CrossRef]
- Wang, J.; Cong, Z.; Huang, X.; Hou, C.; Chen, W.; Tu, Z.; Huang, D.; Liu, Y. Soliseptide A, a cyclic hexapeptide possessing piperazic acid groups from Streptomyces solisilvae HNM30702. Org. Lett. 2018, 20, 1371–1374. [Google Scholar] [CrossRef]
- Tan, Y.; Deng, W.; Zhang, Y.; Ke, M.; Zou, B.; Luo, X.; Su, J.; Wang, Y.; Xu, J.; Nandakumar, K.S.; et al. A marine fungus-derived nitrobenzoyl sesquiterpenoid suppresses receptor activator of NF-kappaB ligand-induced osteoclastogenesis and inflammatory bone destruction. Br. J. Pharmacol. 2020, 177, 4242–4260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, W.; Zeng, J.; Meng, J.; Jiang, H.; Wang, J.; Xing, D. Niemann-Pick C1-Like 1 inhibitors for reducing cholesterol absorption. Eur. J. Med. Chem. 2022, 230, 114111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Song, Z.; Wang, X.; Xue, J.; Xing, D. One-step modification to identify dual-inhibitors targeting both pancreatic triglyceride lipase and Niemann-Pick C1-like 1. Eur. J. Med. Chem. 2021, 216, 113358. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Y.; Eelen, G.; Zhou, Q.T.; Feng, W.B.; Labroska, V.; Ma, F.F.; Lu, H.P.; Dewerchin, M.; Carmeliet, P.; et al. A high-throughput screening campaign against PFKFB3 identified potential inhibitors with novel scaffolds. Acta Pharmacol. Sin. 2022, 44, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, G.Q.; Zhou, Y.; Lee, W.J.; Buchanan, C.M.; Denny, W.A.; Rewcastle, G.W.; Kendall, J.D.; Dickson, J.M.J.; Flanagan, J.U.; et al. High-throughput screening campaigns against a PI3Kalpha isoform bearing the H1047R mutation identified potential inhibitors with novel scaffolds. Acta Pharmacol. Sin. 2018, 39, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.; Courtney, K.; Andres, V.; Featherstone, R. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).