1. Introduction
Trauma, cancer, and congenital disorders result in bone lesions and defects [
1]. Consequently, several million people worldwide undergo bone grafting yearly, which has a high economic impact on health systems [
2]. There are three main types of bone grafts: autografts, allografts, and xenografts; however, they all possess limitations (i.e., donor site morbidity, immunogenicity, biological variability, low availability), which affect their use. Due to these limitations, alternative man-made and natural bone substitute materials are used clinically for bone regeneration [
3]. Calcium carbonate, calcium sulfate, hydroxyapatite and tricalcium phosphate have all been used as bone graft substitutes [
4]. Porous hydroxyapatite is one of the most frequently used materials and has been shown to promote in vivo osteoblastic differentiation whilst being osteogenic and osteoconductive [
5,
6]. This material is mildly osteoinductive. However, this is usually associated with using growth factors such as bone morphogenetic proteins. Hydroxyapatite bone graft substitutes have a low biodegradation rate, which is not always compatible with the growth of new bone [
7]. As bone formation progresses on the surface of the graft, the ability of a material to degrade and be replaced by bone tissue is important. Tailored resorption of synthetic bone graft material can induce and improve the capacity of bone required for the repair of fractures by enabling the material to be assimilated with the surrounding living bone tissue. The discovery in 1931 of perfectly Osseo integrated teeth made of nacre (the inner layer of mollusc shells) in Mayan jaw bones encouraged researchers to explore the osteogenic and osteoinductive potential of the mother of a pearl [
8]. Marine bivalve shells have a nacre lining that comprises anhydrous calcium carbonate plates cemented by a thin organic matrix (1–5% wt.) [
9]. This material has the potential to be used as a bone graft substitute.
Anhydrous CaCO
3 exists in three crystalline polymorphs: calcite, aragonite and valerite. Calcite is the most stable and least soluble in water and is found in a trigonal crystalline state in nature. Valerite, on the other hand, is the least stable, exists in a hexagonal crystalline form, and is present in bird eggs and many marine living organisms, such as gastropods and mollusc pearls [
10]. Aragonite occurs in the orthorhombic form, commonly found in seashells (e.g., bivalves) and corals. Aragonite is extraordinary biocompatible [
11], is denser than calcite, and can be integrated and replaced by bone [
12]. The biogenic aragonite in the nacre makes the shells strong enough to protect the shellfish from harsh environments, and the nacre has the potential to be used as a biomaterial with outstanding mechanical properties. The fracture toughness of nacre is 3000 times greater than that of pure aragonite [
13], and its mechanical properties are comparable to titanium [
14]. Several published articles have suggested that the bone response to nacre combined with its strength makes it a promising candidate for dental implants, whilst nacre powder has been used to reconstruct maxillary defects [
8,
15,
16].
Furthermore, calcium carbonate is more soluble than hydroxyapatite, with a faster resorption rate. Finally, biogenic CaCO
3, derived from waste shells or invasive species such as the slipper limpet, would reduce environmental impact and be more economical than a bone graft substitute fabricated from chemicals obtained from limestone [
10,
17]. Although extensive research has been conducted on the bioactivity, biocompatibility, and osteoinductive properties of nacre from various species such as
Hyriopsis cumingii,
Arctica islandica,
Pinctada Maxima,
Pinctada margaritifera,
Limnoperna fortunei,
Anadara granosa, and pure aragonite from cuttlefish skeletons (
Sepia officinalis) and corals (
Porites sp.) [
18,
19,
20,
21,
22], shells of
C. fornicata have yet to be studied as a potential biomaterial for bone regeneration.
The slipper limpet, or
C. fornicata, is a marine gastropod native to eastern North America but invasive to UK and EU waters. It has displaced native species and grown to harmful numbers. Not being a commercial species,
C. fornicata has no commercial value. However, it is regularly caught as a by-catch of oyster and scallop dredging, often illegally dumped above high tide to naturally deteriorate. In the UK, it is illegal to return slipper limpets to the water or seabed [
23]. This invasive species could offer a novel, cost-effective material for bone graft substitutes, incentivizing activities for cleaning up this invasive species whilst providing environmental improvements [
16]. These opportunities, together with the increasing demand for large volumes of inexpensive biomaterials from natural resources for the repair of bone defects [
24], have led us to investigate the osteogenic potential of this new source of aragonite to produce bone graft substitutes and structural biomaterials made from the mantle of
C. fornicata.
This study evaluated, for the first time, the ability of the mantle of C. fornicata to enhance the proliferation and differentiation of human adipose-derived stem cells into osteoblasts (OBs).
3. Discussion
Bone tissue engineering aims to produce biomaterials that can substitute and induce the regeneration of damaged tissue. These materials require either the introduction of cells capable of reconstructing the tissue or the recruitment of such cells from local or systemic sources, such as mesenchymal stem cells and osteoblasts [
27,
28].
In the current in vitro study, we investigated the physicochemical characteristics of slipper limpet discs and their ability to support the attachment, spreading and osteogenic differentiation of hAdMSCs. This work has sought to identify a novel biomaterial for industrial use with a low ecological and economic impact.
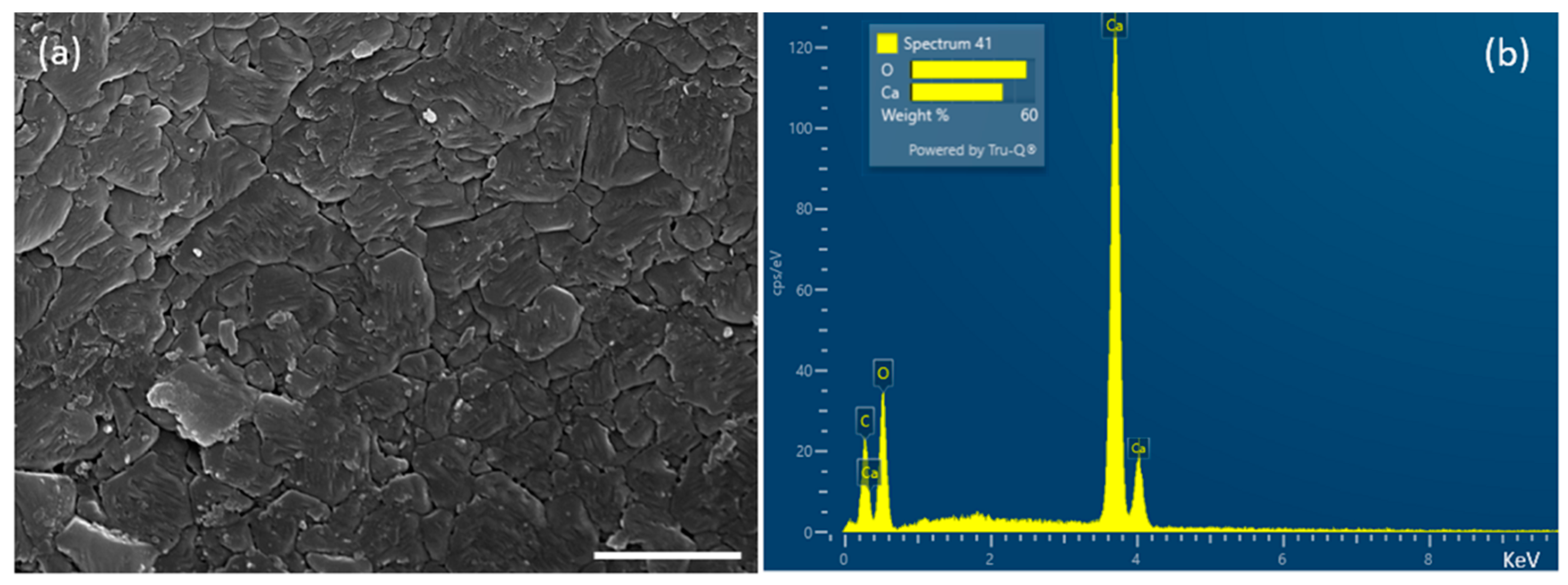
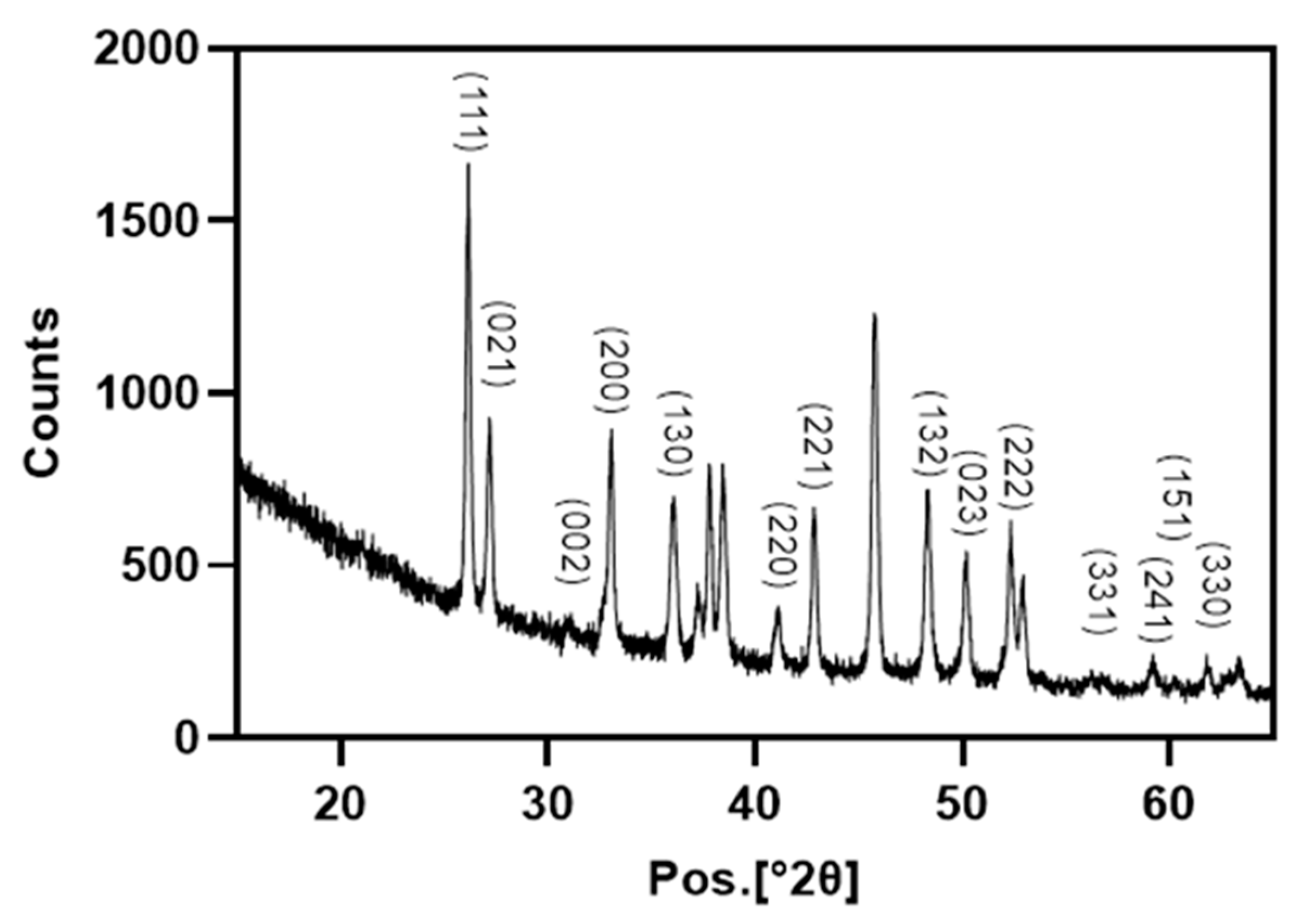
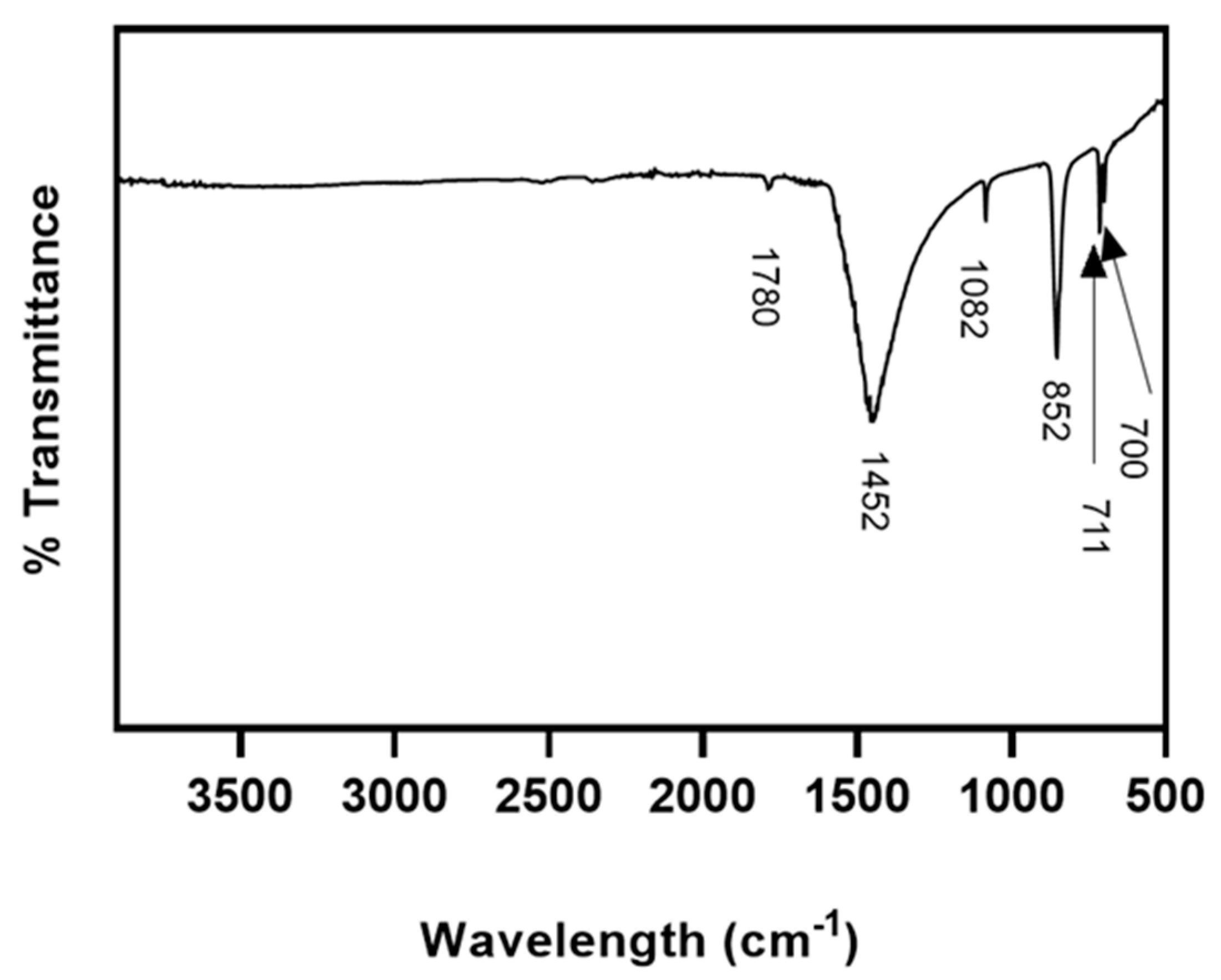
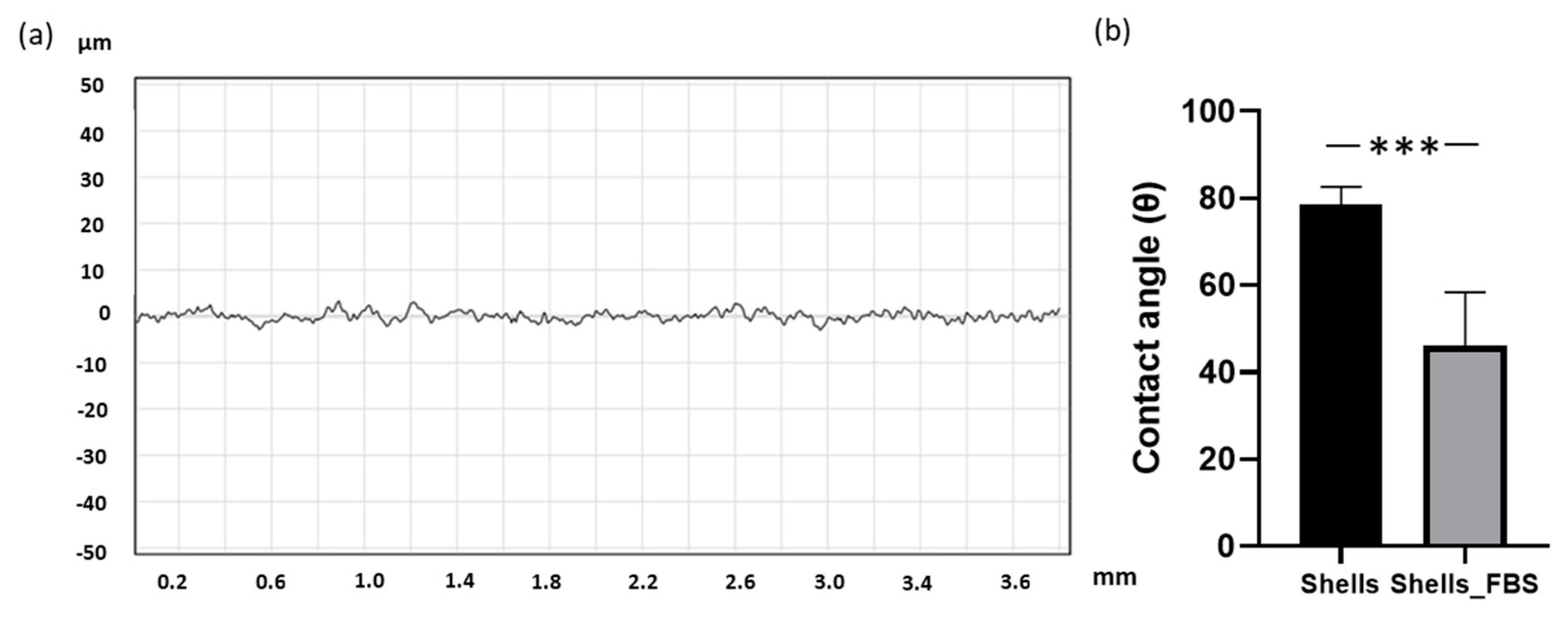
In our study, XRD and FT-IR showed that limpet shells were composed mainly of calcium carbonate in the form of aragonite. These results are consistent with Batzel et al. where XRD using powdered
C. fornicata shells showed they were made of the calcium carbonate polymorph aragonite with no indication of the presence of other crystalline minerals [
29]. In addition, we observed that the surface of the shell naturally exhibited a uniform micro-roughness that was slightly hydrophilic. Hydrophobicity and roughness are crucial surface properties for interaction of a biomaterial with the biological environment, including tissues and cells [
24]. Surface topography is known to influence cell adhesion, proliferation, and differentiation. For example, Faia-Torres et al. demonstrated that, on polycaprolactone surfaces, gradients with micrometer-scale roughness supported greater osteogenesis than sub-micrometer surfaces [
25]. Similar results were reported by Li et al. on titanium surfaces, where the osteogenesis of human osteoblastic SaOS-2 cells was enhanced with increased surface roughness (from approximately 100 to 400 nm) [
30]. However, Davison et al. have shown that in vivo, beta-tricalcium phosphate (TCP) graft substitutes with a submicron-scale surface induced significantly improved bone formation than microstructured TCP [
31]. The interfacial free energy of the implant surface is another physicochemical property that influences its interaction with biological environments. Generally, the higher the surface energy, the higher the hydrophilicity, and the higher the cell adhesion. It has been attributed to the rapid spreading of serum on the surface, which provides an overall coating of bioactive factors that can influence early cell adhesion, proliferation, and differentiation [
32].
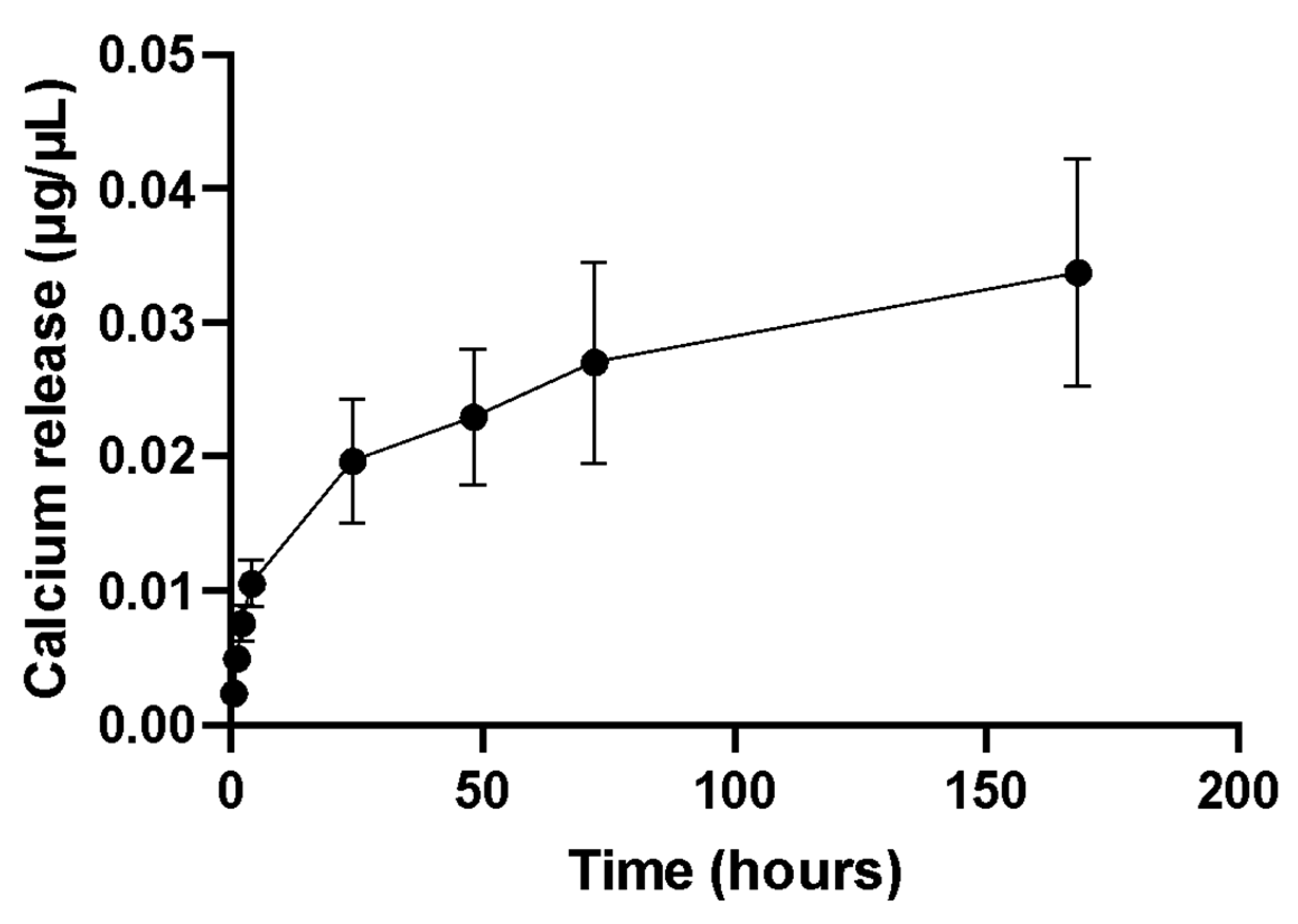
In our work,
C. fornicata discs, incubated in PBS, could release Ca
2+ over the experimental period of 7 days. Ca
2+ in the fluid surrounding cells plays a key role in osteoinduction. It has been shown that the differentiation of human bone marrow-derived MSC towards osteoblasts is accompanied by the expression of Ca
2+ binding proteins [
33].
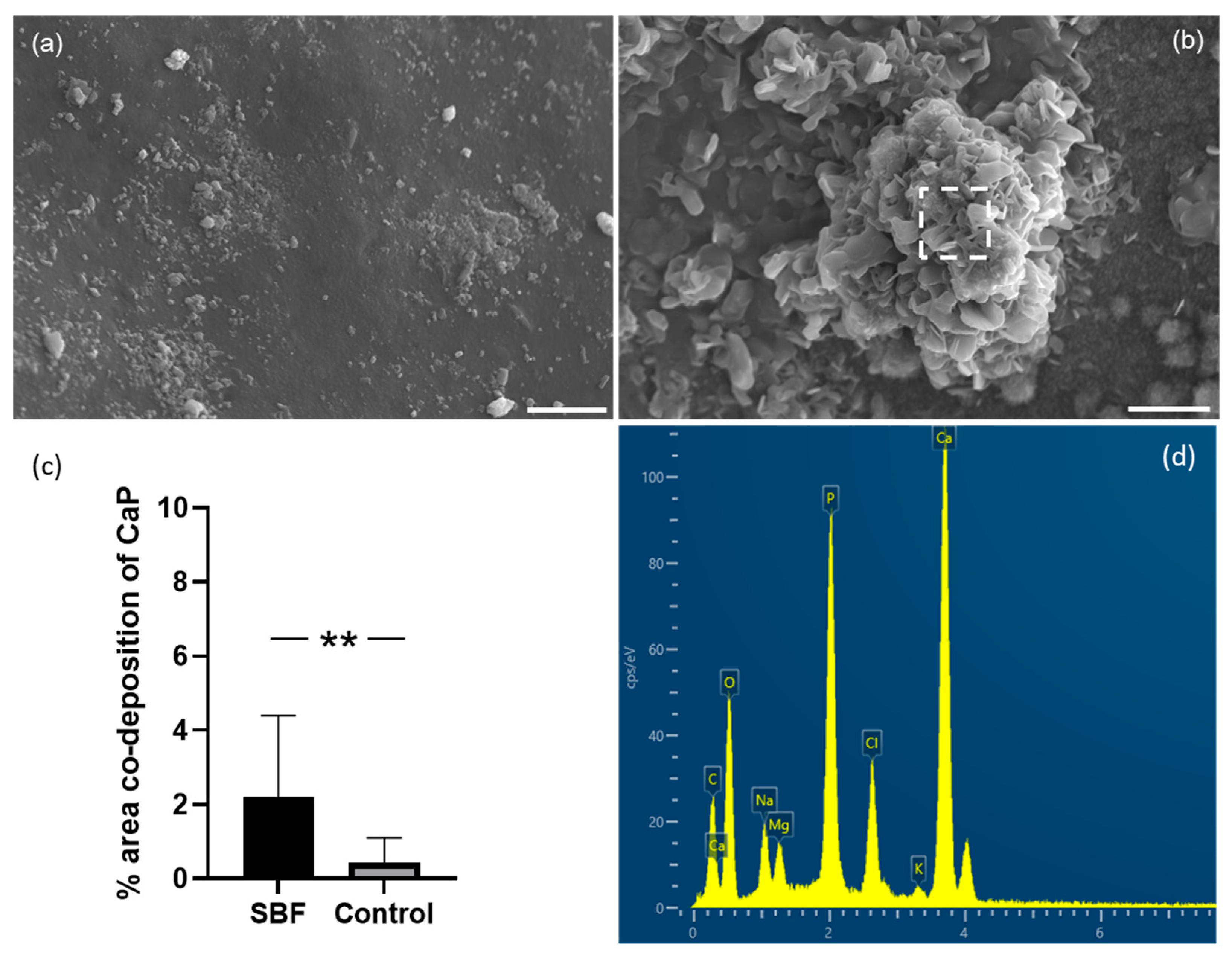
Osteotransduction, which refers to transforming a biomaterial into new bone tissue after implantation, is a property of calcium phosphate biomaterials. Hydroxyapatite (HAP) formation on the surface is desirable for an implant because HAP can form strong bonds with natural bone. We, therefore, assessed whether discs made from the mantle of
C. fornicata could promote the formation of calcium phosphate on their surface. We showed that
C. fornicata discs, immersed in SBF for 21 days, presented Ca/P deposits on their surface [
21,
34]. In addition, the shell surface was partially covered by spherical nodules composed of calcium phosphate, as shown in EDS. The level of 4% is the area that was associated with both calcium and phosphate. Although the areas occupied by the nodules are not large, the results show that the shell surface still induces osteoinduction.
Increasing the surface area of the aragonite by roughening it or by making it porous would potentially increase calcium release and enhance hydroxyapatite deposition. Interestingly, in our study, the calcium-to-phosphorus ratio was 1.69, which is very similar to the ideal ratio for natural hydroxyapatite, 1.67 [
26]. There are two main theories to explain why aragonite, and in particular nacre, can promote hydroxyapatite deposition. One theory is that nacre has an organic component performing a function similar to collagen in bone and promoting hydroxyapatite formation [
26]. Alternatively, Ni and Ratner hypothesized that the nacre surface is activated after calcium ions are released from the shells [
35]. The free calcium ions bind to phosphate ions and precipitate on the nacre surface as hydroxyapatite. Calcium ions do not re-precipitate as calcium carbonate because the solubility of HAP is much lower than that of aragonite. As seen in our study, when immersed in SBF, calcium ions were released from the
C. fornicata discs immediately and this continued for seven days, which was the duration of the experiment. Overall, our results suggest that
C. fornicata discs have the potential to be a valuable biomaterial for bone tissue engineering owing to their ability to release calcium ions and promote the formation of hydroxyapatite on their surface and this promotes osteoblastic differentiation of stem cells.
In this study, human adipose-derived MSCs were used to investigate osteogenesis, as they have the potential to differentiate into osteoblasts [
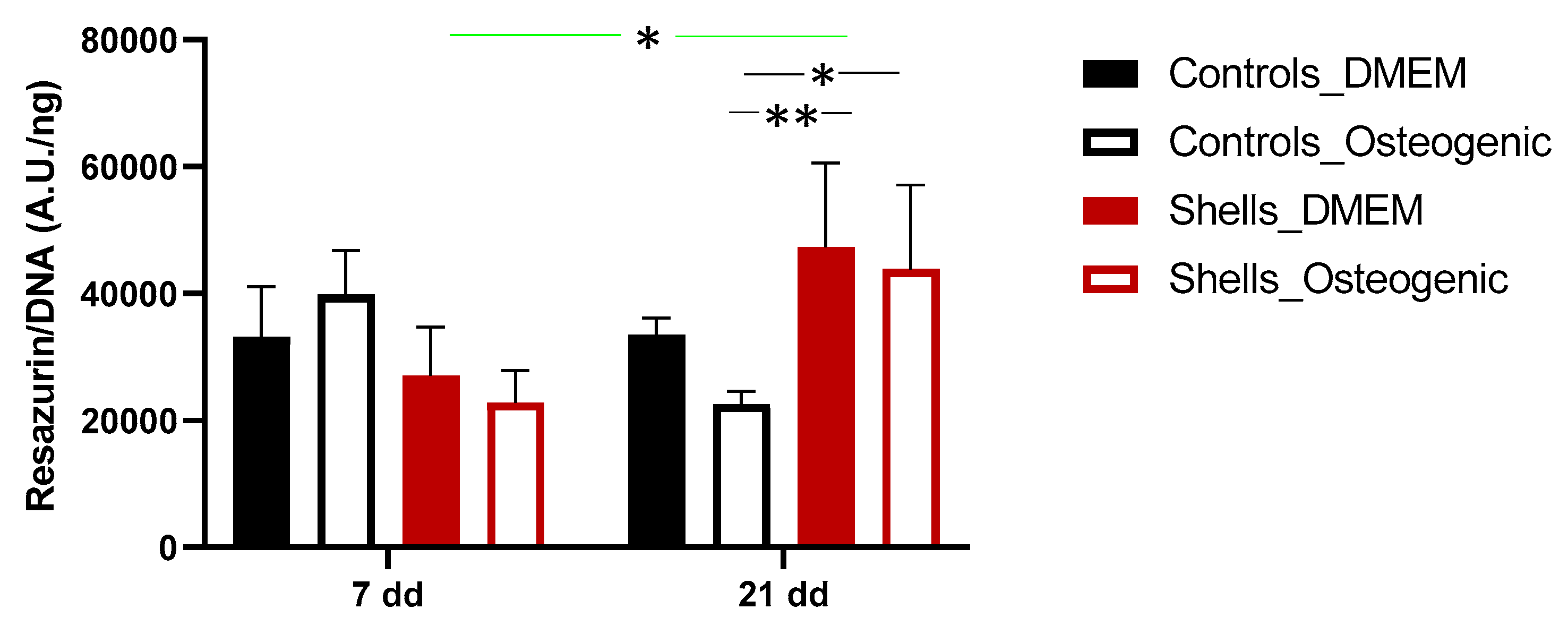
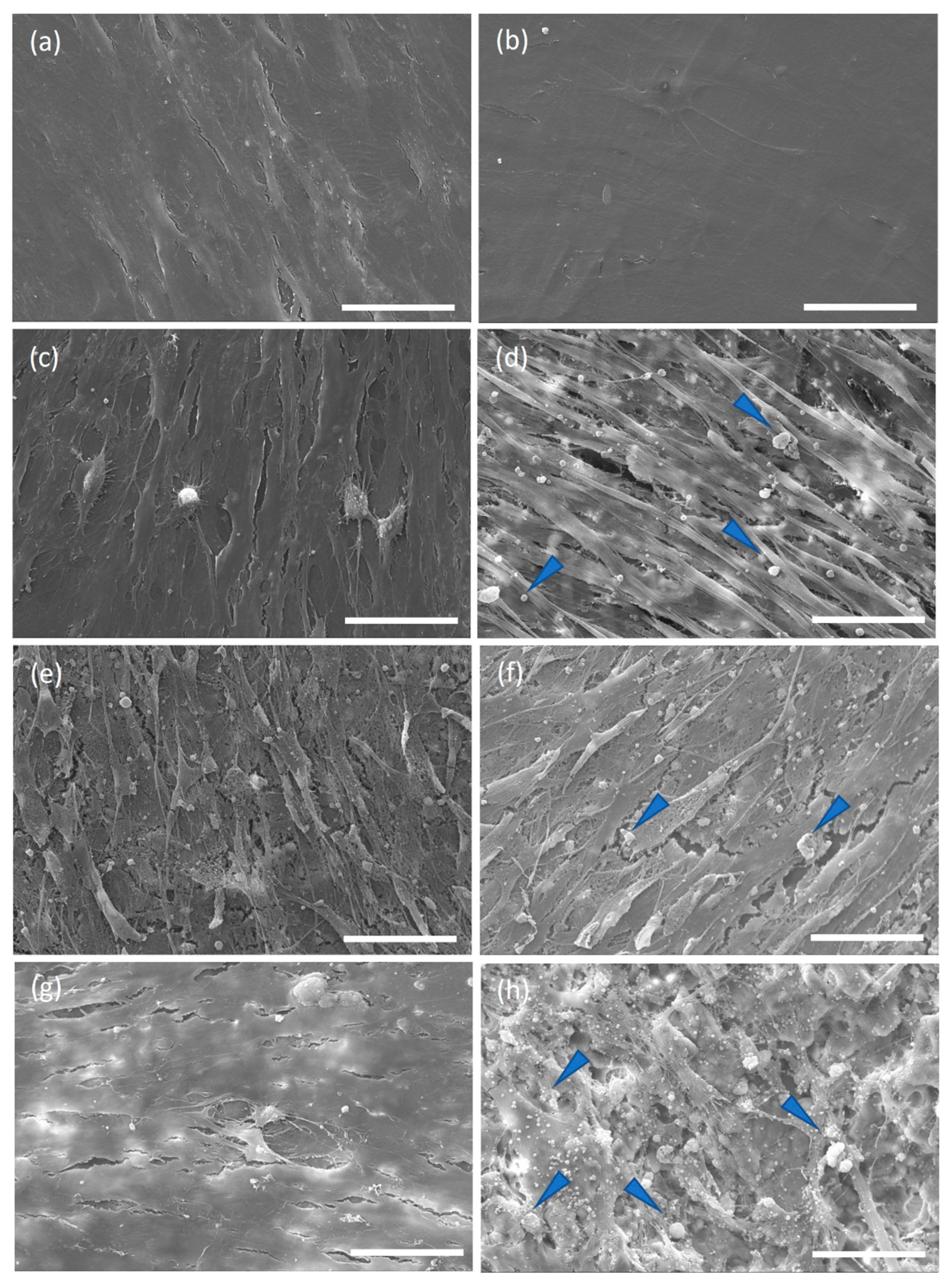
36]. The fluorescence quantification of resazurin and the SEM images demonstrated that the cells could attach and proliferate on discs from shells, as previously described for hAdMSC grown on other shell surfaces [
21]. Interestingly, despite the cell attachment efficiency of 38% after one day on shell surfaces compared to the coverslips, by day 7, the proliferation was similar. This suggests that hAdMSC proliferated faster on
C. fornicata than on the coverslip surface. Similar observations were made by Kamba and Zakaria using hFOB 1.19 co-cultured with calcium carbonate nanocrystals derived from cockle shells [
37]. In our study, cells grown on both materials presented a spindle-like shape on days 7 and 21. Furthermore, the mineral deposition and the extracellular matrix production by cells on the shells in a non-osteogenic medium indicate that the shells promoted osteoblastic differentiation.
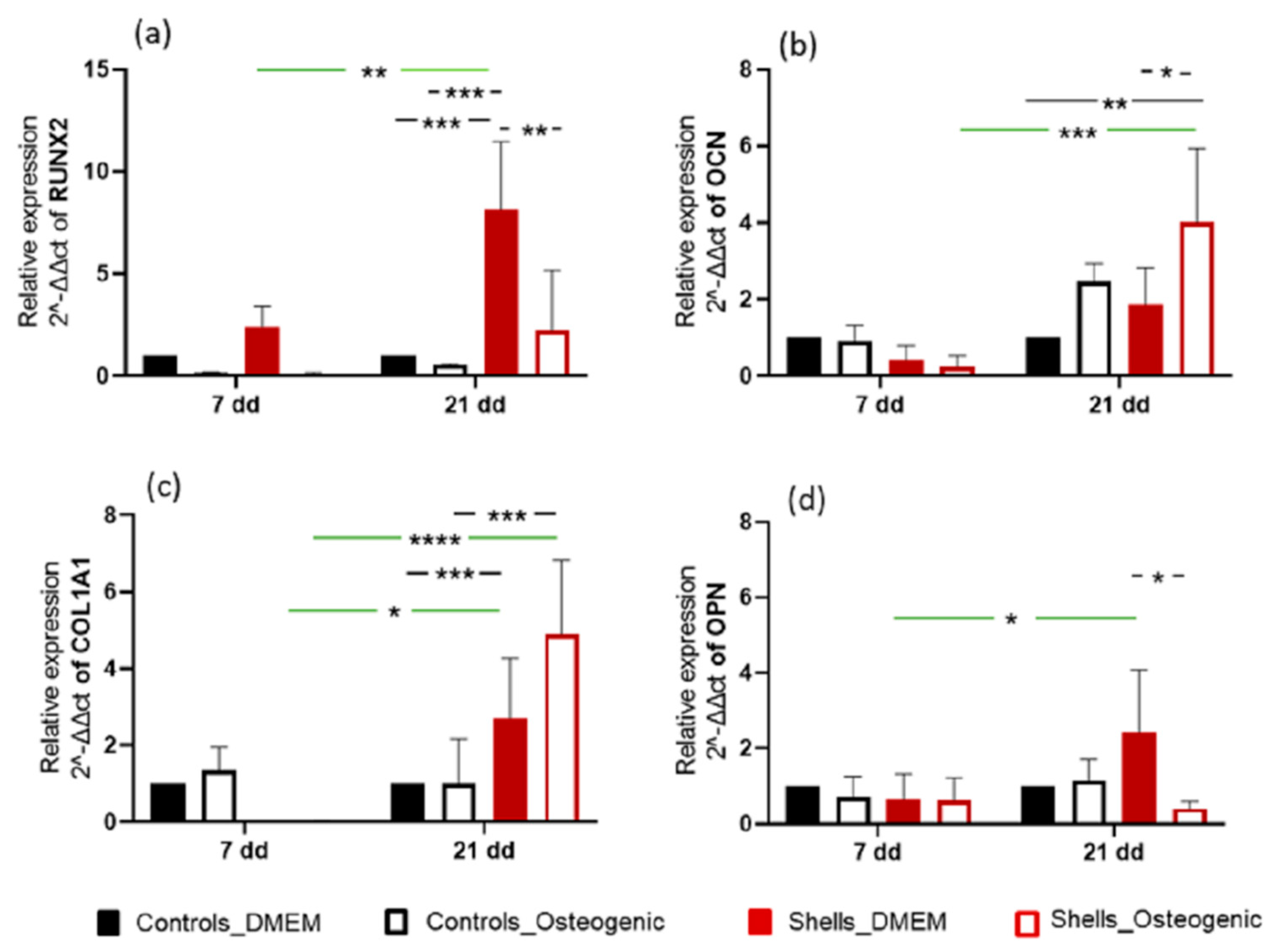
We quantified the relative gene expression of early (RUNX2 and Col1A1) and late bone-specific markers (OPN and OCN) to confirm osteogenic differentiation. These early and late bone-specific markers have been well-documented in previous reports [
38]. The cells used for this study were a multipotent human adipose-derived stem cell line, which can differentiate into different cell lineages, such as adipocytes, myocytes, chondrocytes, and osteoblasts. In vivo, differentiation depends on the composition and 3D organisation of the extracellular matrix (ECM) and the combined effect of growth factors and other morphogens in their immediate environment [
39]. In our in vitro study, osteogenic differentiation was initiated by the shell. RUNX2 is essential for osteogenic differentiation and regulates the temporal expression of other osteogenic markers and the secretion of the extracellular matrix. COL1A1 expression was higher at 21 days than at seven days, for the cells grown on
C. fornicata shells, both with and without an osteogenic medium. Osteocalcin expression was significantly greater when cells were grown on
C. fornicata shells indicating that the shells promoted gene expression related to osteogenic differentiation. RUNX2 upregulation was not recorded for cells grown on
C. fornicata in complete medium but was when the osteogenic medium was used. Nevertheless, the effect of growing cells on the shell surface was to promote mineralization even when incubated in DMEM without an osteogenic medium.
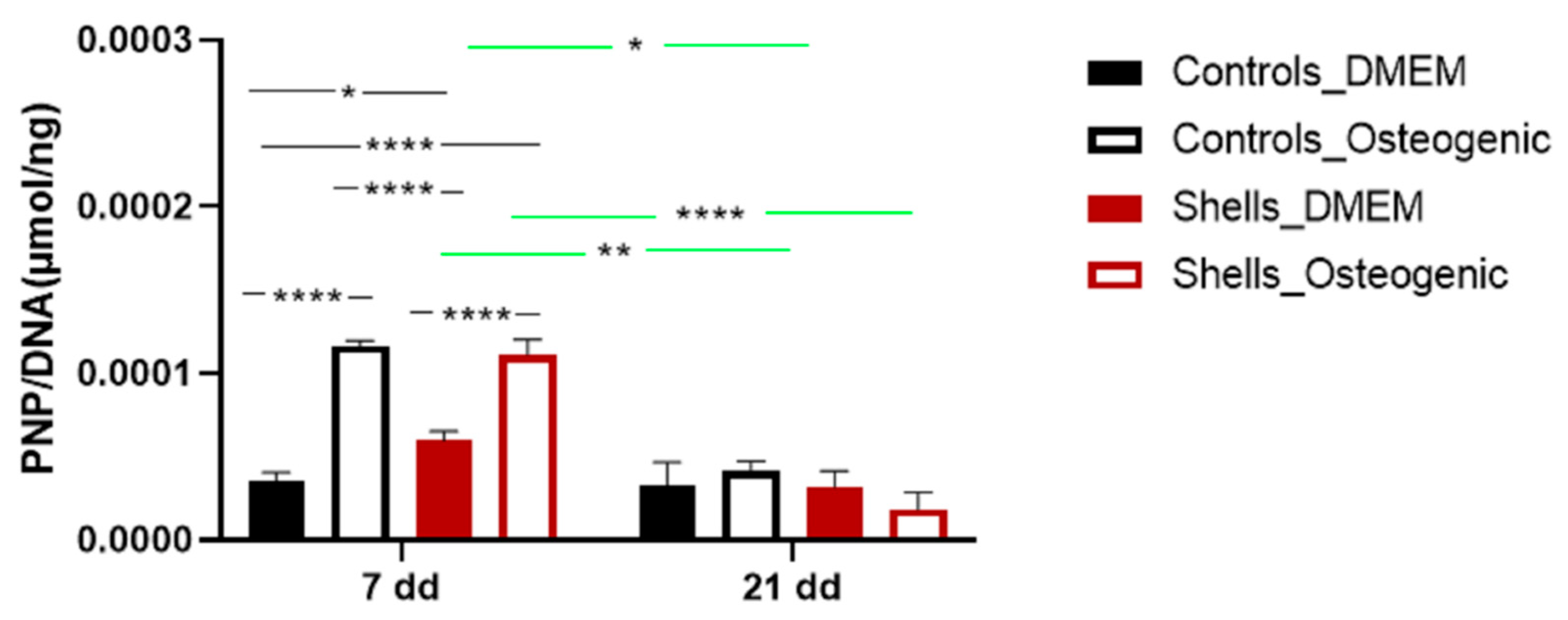
To evaluate markers of osteogenesis, we also examined the activity of the alkaline phosphatase (ALP), an early and transient indicator of osteogenic differentiation. ALP was present in all cells on day 7. The ALP activity was significantly higher for cells grown on shells without an osteogenic medium. Adding osteogenic medium at day 7 increased the levels of ALP for cells on coverslips and on shells suggesting that the osteogenic media suppressed the surface effect. Corrêa de Almeidaa et al., working with
Limnoperna fortunei shells (also predominantly aragonite), didn’t find any increase of ALP activity in human adipose stem cells grown for five days in DMEM medium, suggesting that
C. fornicata shells may be better promoters of bone formation [
21]. However, when Corrêa de Almeidaa et al. powdered the same shells, there was a significant increase in ALP activity due to the larger contact surface, which considerably increased calcium release in the media, favoring a faster osteogenic process. In our study, ALP activity diminished for all groups on day 14 except for the control group. The decline of ALP activity might be associated with the rapid formation of minerals. ALP is required before the onset of matrix mineralization, providing localized enrichment of inorganic phosphate, one of the components of apatite [
40]. In our study, SEM-EDS images showed mineralization at day 7, indicating that earlier time points for ALP measurement may have shown an even larger response to the shell surface.
The results suggest that the shell composition is partially responsible for the mineralization of the extracellular matrix, as shown by the differentiation of the stem cells and the hydroxyapatite deposition. Furthermore, our results indicate that C. fornicata shells (regardless of using osteogenic medium) promote bone formation. Although the shell material would need to be fabricated into a bone graft substitute material which would depend on the application, its availability and relatively low cost make it an attractive option with a low carbon footprint. Future studies should focus on using granular material derived from shells and possibly developing a porous scaffold made from shells to mimic bone’s porous interconnected network which facilitates vascularization and rapid growth of newly formed bone.
4. Materials and Methods
4.1. Preparation of Samples
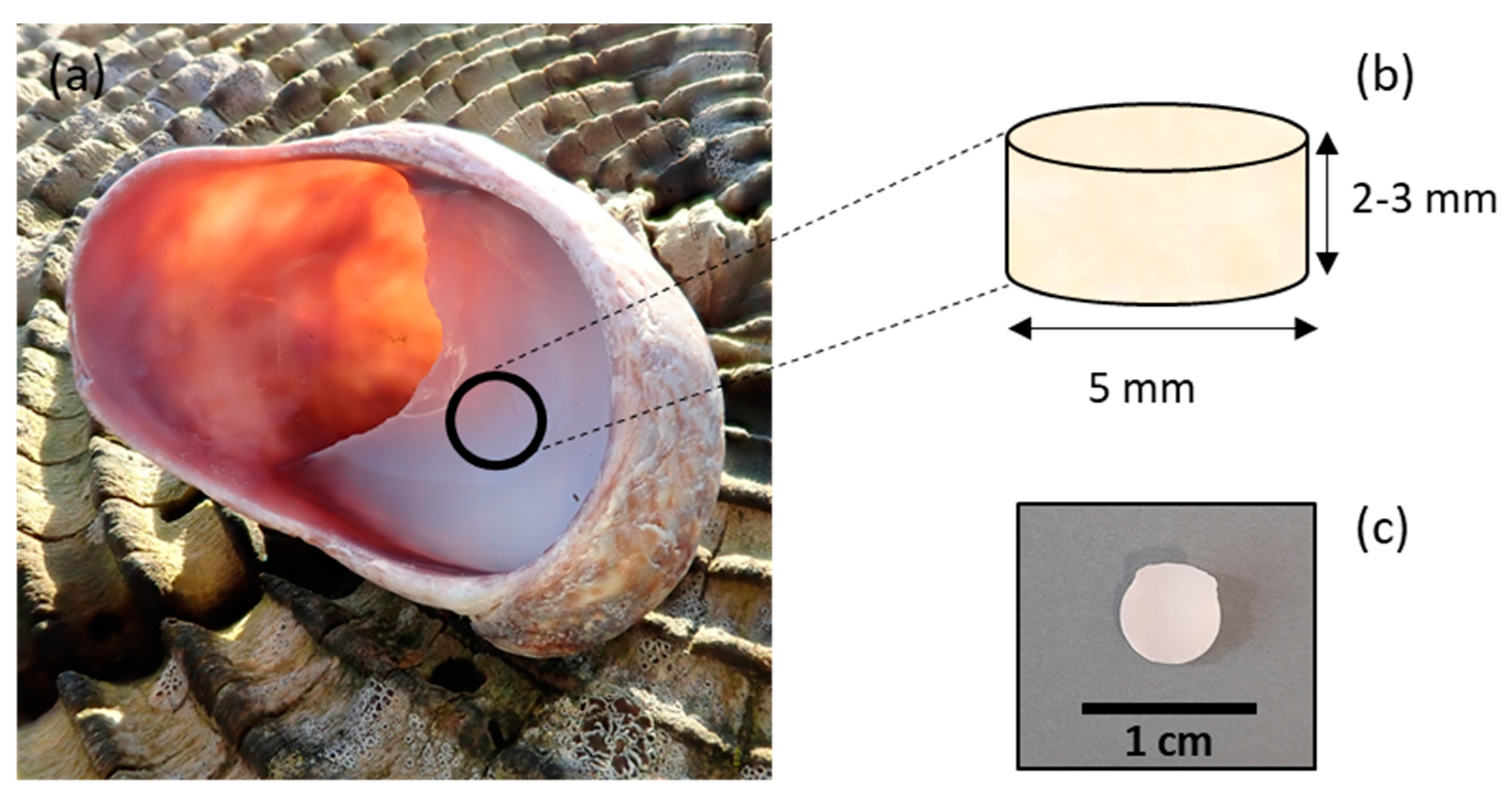
C. fornicata shells were collected in Pembrokeshire, Wales, UK. The samples were rinsed with distilled water. The samples were then air-dried at room temperature. Shells were shaped as cylinders by coring the mantle of the shell with a 5 mm diameter diamond core fitted into a pillar drill (Sealey, Suffolk, UK) (
Figure 11).
4.2. FT-IR
C. fornicata powder was obtained by grinding the shells to a fine powder in a ceramic mortar. FT-IR spectra of C. fornicata powder were recorded using a Varian FT-IR 640-IR Instrument (Agilent, Santa Clara, CA, USA) and spectra were processed using Agilent Resolution Pro software (Agilent Technologies, Mulgrave, Australia).
4.3. XRD
C. fornicata powder was prepared as described above. X-ray diffraction was performed using a X’ pert3 powder X-ray diffractometer (Malvern PANalytical B.V., Almelo, The Netherlands). The dataset was collected using Cu Ka radiation in the 2θ range of 5 to 89 at a scan speed of 0.006°2θ/min. The generator settings were 35 mA and 40 kV.
4.4. SEM and EDS
Scanning electron microscopy of shells coated with gold/palladium (Au:Pd 80:20) using a Quorum 150 TES sputter coater was undertaken by a Tescan MAIA 3 scanning electron microscope (15 kV). For SEM-EDS analysis, the specimens were coated with an automatic SEM carbon coater (Agar Scientific, Stansted, UK). Then the samples were imaged by an EVO MA10 SEM (Carl Zeiss, Cambridge, UK) fitted with a LaB6 electron source. Elemental composition was determined by X-ray spectroscopy maps (20 kV) collected using an Oxford Instrument X-Max 80 detector.
4.5. Calcium Release Studies
The discs were rinsed with water, autoclaved, and finally soaked in 1 mL of PBS (without calcium and magnesium) (Fisher, Loughborough, UK) in sterile tubes. The samples were incubated at 37 °C with shaking (100 rpm). At scheduled time points (0.5, 1, 2, 4, 24, 28, 72 and 168 h), 0.5 mL of solution was taken and replaced with the same volume of fresh PBS. The concentration of calcium was determined with a calcium colorimetric kit (Merck Life Science, Gillingham, UK) according to the manufacturer’s instructions. Briefly, 50 μL of samples were mixed with 90 μL of chromogenic reagent and 60 μL of calcium assay buffer. Then, the plates were incubated at room temperature for 10 min, and the absorbance was read at 575 nm (SpectraMax i3x) (Molecular Devices, San Jose, CA, USA), using PBS as a blank. The experiment was conducted in triplicate.
4.6. Roughness Analysis
The surface roughness of the C. fornicata discs was measured using a Formtracer SV-C3200 instrument and analysed using the FORMTRACEPAK software (Mitutoyo UK Ltd., Andover, UK). The samples were kept steady using blue tack. The measured length was 4 mm, the measuring speed was 1.00 mm/s, the measuring pitch was 0.0005 mm, and the Z1-axis range was 800 μm. The measurements were repeated three times per sample, and the experiment was done in triplicate.
4.7. Wettability
Contact angle measurements for each FBS-coated (made as described in paragraph 4.9) and uncoated disc were performed using an optical contact angle meter CAM101 (KSV Instruments Ltd., Espoo, Finland). The measurements were conducted with deionized water and were repeated three times for each surface.
4.8. In Vitro Bioactivity
The deposition of apatite on the surface of
C. fornicata from simulated body fluid was assessed according to the methods of Kokubo and Takamada [
20]. Sodium chloride (NaCl), sodium hydrogen carbonate (NaHCO
3), potassium chloride (KCl), di-potassium hydrogen phosphate trihydrate (K
2HPO
4.3H
2O), magnesium chloride (MgCl
2), calcium chloride (CaCl
2), sodium sulfate (Na
2SO
4), tris-hydroxymethyl aminomethane ((HOCH
2)
3CNH
2) and hydrochloric acid were bought from Fisher (Loughborough, UK). The autoclaved samples were soaked in 15 mL of SBF in 50 mL tubes. Samples were incubated at 37 °C for 21 days. Samples kept in deionized water were used as a control. The SBF was changed every 3–4 days. Finally, the samples were gently washed in deionized water and vacuum dried. Samples were carbon-coated and imaged as described above. EDS images for calcium (Ca) and phosphorus (P) were analyzed using ImageJ (NIH, Madison, WI, USA) after being overlaid to corresponding SEM images. The total area of overlapping for Ca and P onto the total sample surface area was used to calculate the percentage of Ca and P salt deposition (
n = 3).
4.9. In Vitro Cell Culture
An adipose-derived stem cell line (Asc52telo, ATCC® SCRC-4000™) was bought from LGC standards (Teddington, UK). The cells tested negative for HIV, HepB, HPV, EBV, CMV and mycoplasma. Cells were maintained, under aseptic conditions, in DMEM (high glucose, GlutaMAX™, pyruvate) supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin (Fisher, Loughborough, UK). The cells were cultured at 37 °C in a 5% CO2 humidified incubator, and the culture medium was changed twice weekly. At 80–90% confluence, the culture medium was removed, and the cells were washed with Dulbecco’s phosphate-buffered saline (DPBS) (Fisher, Loughborough, UK). The cells were detached following the incubation with 0.25% trypsin-EDTA (Fisher, Loughborough, UK) at 37 °C. Cells between passages 4 and 5 were used for the study.
4.10. Cell Seeding on Discs
Discs were autoclaved and then put in 48 (low cell attachment) well plates. Discs were coated with FBS for 4 h and air-dried, under laminar flow, overnight. Human adipose-derived stem cells (60,000) were seeded in 20 μL of complete medium were seeded on each sample and control (12 well plates). More precisely, 30,000 cells in 10 μL were seeded on one side of the disc and then incubated for 90 min. The same was repeated for the opposite side of the scaffold. Then, the samples were covered with a complete medium and incubated (5% CO2 and 37 °C). After one day, the discs were transferred to a new plate, covered with either a basic or osteogenic medium (StemProTM Osteogenesis differentiation kit) (Fisher, Loughborough, UK), and cultured for 21 days.
4.11. Cell Attachment and Proliferation
After 24 h since seeding, a resazurin assay (Merck, Gillingham, UK) was performed. A filter sterile 0.1 mM resazurin salt solution in a complete medium was added to each sample. The plates were incubated (5% CO2 and 37 °C) for 2 h. The working solution, incubated in the same conditions, worked as a blank. Then, 100 µL aliquots were transferred to a new 96-well plate, and the fluorescence was immediately read using a plate reader (ex/em 540/585 nm). The experiment was performed in triplicate (n = 3). At the end of the experiment, the resazurin solution was removed, and a fresh medium was added.
4.12. Alkaline Phosphatase Activity (ALP)
Carbonate lysis buffer (450 μL) (1% Triton in 0.2 M carbonate buffer, pH 10) (Merck, Gillingham, UK) was added to the shells and controls. Afterwards, the extracts were frozen/thawed thrice and lysed using a 21 G needle. For ALP quantification, 50 μL of lysate was mixed with 100 μL of p-Nitrophenyl Phosphate (pNPP, 6 mM) carbonate solution (containing 10 mM MgCl2) (Merck, Gillingham, UK). The plate was incubated at 25 °C for 1 h. Absorbance was read at 405 nm. Data were normalised against DNA. DNA quantification was done using ab156902 General DNA Quantification Kit (SYBR Green) (Abcam, Cambridge, UK), according to the manufacturer’s instructions. More precisely, 5 µL of extracts were mixed with 100 µL of 1X DNA assay solution and the fluorescence was read, after 10 min, (480/520 ex/em. The specific activity of cells was expressed as μmoles/ng DNA (±SD).
4.13. Cell Morphology by SEM
On days 7 and 21, samples were fixed in 2.5% glutaraldehyde (Merck, Gillingham, UK) in 0.1 M cacodylate (Merck, Gillingham, UK) overnight at 4 °C. The samples were then washed with deionized water three times and dehydrated with an ethanol series (20 min each, 50%, 70%, 90% and 100%) (Fisher, Loughborough, UK) and with 1:2 and 2:1 HMDS (hexamethyldisilazane): EtOH and 100% HMDS (overnight) (Merck, Gillingham, UK). The samples were then mounted on stubs and gold-coated (Polaron e500) (Quoram Technologies, Laughton, UK) before imaging at 20.00 kV by SEM.
4.14. RNA Extraction and RT-qPCR
Qiazol lysis reagent (Qiagen, Manchester, UK) was used to lyse cells at 7 and 21 days. Cell content was homogenised using a sterile 25-gauge needle and centrifuged at 18,000×
g for 2 min at room temperature. The supernatant was processed using a RNeasy PLUS micro kit for RNA isolation (Qiagen, Manchester, UK). RNA was quantified using a nanodrop (Nanodrop ND-1000) (Thermo Fisher Scientific, Wilmington, NC, USA) and stored at −80 °C until use. Complementary DNA (cDNA) was obtained from RNA (250–500 ng from each sample) using High-Capacity cDNA Reverse Transcription Kit (Fisher, Loughborough, UK) on a thermal cycler (T100) (Biorad, Watford, UK), according to the manufacturer’s instructions. RTq-PCR was performed in a LightCycler96 (Roche, Burgess Hill, UK), using SSO Universal SYBR Green Supermix (Fisher, Loughborough, UK), and data were analysed using the LightCycler SW 1.1 analysis software (Roche, Penzberg, Germany). Relative gene expression was calculated using the comparative 2
−ΔΔCt (expressed as a fold change). GAPDH was used as a housekeeping gene. The primers used were GAPDH (housekeeping gene), RUNX2, OPN, OCN and COL1A1 (Eurofins Genomics, Ebersberg, Germany) (
Table 1).
4.15. Statistical Analysis
Data was parametric, so statistical significance was analysed using the unpaired t-test and ANOVA and all results are shown as mean ± SD. Samples were run in triplicate for the biochemical assays. Statistical analysis was performed using GraphPad Instant Software (GraphPad Software Inc., Boston, MA, USA). All experiments were repeated at least three times.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}