
Petrosamine Revisited. Experimental and Computational Investigation of Solvatochromism, Tautomerism and Free Energy Landscapes of a Pyridoacridinium Quaternary Salt


Abstract
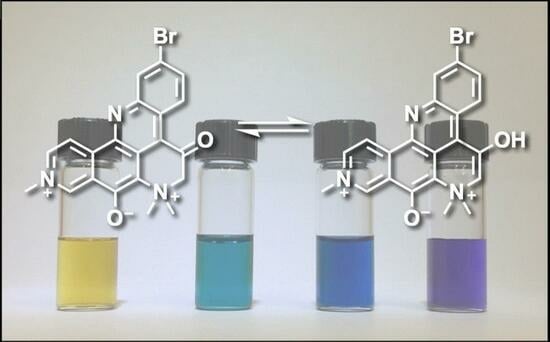
1. Introduction
2. Results and Discussion
2.1. QM Calculations
2.2. pKa of Petrosamine (1), Does the Structure of 1 Exhibit Substantial Enol Content?
2.3. Kinetic Measurements of Hydrogen-Deuterium Exchange of 1
3. Materials and Methods
3.1. General Experimental Procedures
3.2. UV-Vis Measurements
3.3. DFT Calculations of 1
3.4. Synthesis of Merocyanine Dye (6b) [16]
3.5. H-D Exchange Measurements of 1 by ESI-TOFMS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Notes
- Molinski, T.F.; Faulkner, D.J. Petrosamine, a Novel Pigment from the Marine Sponge Petrosia sp. J. Org. Chem. 1988, 53, 1341–1343. [Google Scholar] [CrossRef]
- Molinski, T.F. Marine Pyridoacridine Alkaloids: Structure, Synthesis, and Biological Chemistry. Chem. Rev. 1993, 93, 1825–1838. [Google Scholar] [CrossRef]
- Schmitz, F.J.; Agarwal, S.K.; Gunasekera, S.P.; Schmidt, P.G.; Shoolery, J.N. Amphimedine, New Aromatic Alkaloid from a Pacific Sponge, Amphimedon sp. Carbon Connectivity Determination from Natural Abundance 13C-13C Coupling Constants. J. Am. Chem. Soc. 1983, 105, 4835–4836. [Google Scholar] [CrossRef]
- Marshall, K.M.; Barrows, L.R. Biological activities of pyridoacridines. Nat. Prod. Rep. 2004, 21, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Nukoolkarn, V.S.; Saen-oon, S.; Rungrotmongkol, T.; Hannongbua, S.; Ingkaninan, K.; Suwanborirux, K. Petrosamine, a potent anticholinesterase pyridoacridine alkaloid from a Thai marine sponge Petrosia n. sp. Bioorg. Med. Chem. 2008, 16, 6560–6567. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Ngo, A.; Quinn, R.J.; Redburn, J.; Hooper, J.N.A. Petrosamine B, an Inhibitor of the Helicobacterpylori Enzyme Aspartyl Semialdehyde Dehydrogenase from the Australian Sponge Oceanapia sp. J. Nat. Prod. 2005, 68, 804–806. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Bugni, T.S.; Harper, M.K.; Sandoval, I.T.; Manos, E.J.; Swift, J.; Van Wagoner, R.M.; Jones, D.A.; Ireland, C.M. Evaluation of Pyridoacridine Alkaloids in a Zebrafish Phenotypic Assay. Mar. Drugs 2010, 8, 1769–1778. [Google Scholar] [CrossRef]
- Choi, D.-Y.; Choi, H. Natural products from marine organisms with neuroprotective activity in the experimental models of Alzheimer’s disease, Parkinson’s disease and ischemic brain stroke: Their molecular targets and action mechanisms. Arch. Pharm. Res. 2015, 38, 139–170. [Google Scholar] [CrossRef]
- Britton, H.T.K.; Robinson, R.A. CXCVIII.—Universal buffer solutions and the dissociation constant of veronal. J. Chem. Soc. 1931, 1456–1462. [Google Scholar] [CrossRef]
- Exchange of 1H-13C for 2H-13C results in dramatic attenuation of the 13C signal due to quadrupolar relaxation, and absence of the heteronuclear nOe, among other effects.
- Brooker, L.G.S.; Heyes, G.H.; Sprague, R.H.; Van Dyke, R.H.; Van Lare, E.; Van Zandt, G.; White, F.L. Studies in the Cyanine Dye Series. XI. The Merocyanines. J. Am. Chem. Soc. 1951, 73, 5326–5332. [Google Scholar] [CrossRef]
- Brooker, L.G.S.; Heyes, G.H.; Sprague, R.H.; Van Dyke, R.H.; Van Lare, E.; Van Zandt, G.; White, F.L.; Cressman, H.W.J.; Dent, S.G. Color and Constitution. X. Absorption of the Merocyanines. J. Am. Chem. Soc. 1951, 73, 5332–5350. [Google Scholar] [CrossRef]
- Other possible resonance forms, for example invoking electron donation from Br, are considered only minor contributors.
- Simpson, W.T. A Mathematical Treatment of the Color of the Merocyanine Dyes. J. Am. Chem. Soc. 1951, 73, 5359–5363. [Google Scholar] [CrossRef]
- Morley, J.O.; Morley, R.M.; Docherty, R.; Charlton, M.H. Fundamental Studies on Brooker’s Merocyanine. J. Am. Chem. Soc. 1997, 119, 10192–10202. [Google Scholar] [CrossRef]
- Minch, M.J.; Sadiq Shah, S. A Merocyanin Dye Preparation for the Introductory Organic Laboratory. J. Chem. Educ. 1977, 54, 709. [Google Scholar] [CrossRef]
- We confirm the solvatochromism of one example, i, by synthesis and its solvent-dependent UV-vis properties. See Supporting Information.
- Hasselbalch, K.A. Die Berechnung der Wasserstoffzahl des Blutes aus der freien und gebundenen Kohlensäure desselben, und die Sauerstoffbindung des Blutes als Funktion der Wasserstoffzahl. Biochem. Z. 1917, 78, 112–114. [Google Scholar]
- Bordwell, F.K.; Harrelson, J.A., Jr. Acidities and homolytic bond dissociation energies of the αC—H bonds in ketones in DMSO. Can. J. Chem. 1990, 68, 1714–1718. [Google Scholar] [CrossRef]
- Bordwell, F.G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Available online: https://organicchemistrydata.org/hansreich/resources/pka/pka_data/pka-compilation-reich-bordwell.pdf (accessed on 8 July 2023).
- Chiang, Y.; Kresge, J.; Wirz, J. Flash-Photolytic Generation of Acetophenone Enol. The Keto-Enol Equilibrium Constant and pKa of Acetophenone in Aqueous Solution. J. Am. Chem. Soc. 1984, 106, 6392–6395. [Google Scholar] [CrossRef]
- Valpuesta, M.; Díaz, A.; Suau, R. Coulteroberbinone, a quaternary isoquinoline alkaloid from Romneya coulteri. Phytochemistry 1999, 51, 1157–1160. [Google Scholar] [CrossRef]
- Guthrie, J.P.; Cossar, J.; Klym, A. pKa values for substituted acetophenones: Values determined by rates of halogenation. Can. J. Chem. 1987, 65, 2154–2159. [Google Scholar] [CrossRef]
- Novak, M.; Marc Loudon, G. Aminolysis of Acetoxystyrenes. The pKa of Acetophenones in Aqueous Solution. J. Am. Chem. Soc. 1976, 98, 3591–3597. [Google Scholar] [CrossRef]
- Steiner, E.C.; Gilbert, J.M. The Acidities of Weak Acids in Dimethyl Sulfoxide. J. Am. Chem. Soc. 1963, 85, 3054–3055. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]
- Graulich, A.; Scuvée-Moreau, J.; Seutin, V.; Liégeois, J.-F. Synthesis and radioligand binding studies of C-5-and C-8-substituted 1-(3, 4-dimethoxybenzyl)-2, 2-dimethyl-1, 2, 3, 4-tetrahydroisoquinoliniums as SK channel blockers related to N-methyl-laudanosine and N-methyl-noscapine. J. Med. Chem. 2005, 48, 4972–4982. [Google Scholar] [CrossRef]
- Molinski, T.F.; Faulkner, D.J. An antibacterial pigment from the sponge Dendrilla membranosa. Tetrahedron Lett. 1988, 29, 2137–2138. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % H2O (v/v) | λmax1 (nm) | ε1 1 | λmax2 (nm) | ε2 1 |
|---|---|---|---|---|
| 0 | 296 | 184,000 | 648 | 24,000 |
| 20 | 290 | 172,000 | 622 | 20,600 |
| 40 | 288 | 161,000 | 604 | 19,400 |
| 60 | 287 | 141,700 | 589 | 16,500 |
| 80 | 282 | 122,252 | 581 | 12,900 |
| 100 | 281 | 68,500 | 570 | 4700 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gartshore, C.J.; Wang, X.; Su, Y.; Molinski, T.F. Petrosamine Revisited. Experimental and Computational Investigation of Solvatochromism, Tautomerism and Free Energy Landscapes of a Pyridoacridinium Quaternary Salt. Mar. Drugs 2023, 21, 446. https://doi.org/10.3390/md21080446
Gartshore CJ, Wang X, Su Y, Molinski TF. Petrosamine Revisited. Experimental and Computational Investigation of Solvatochromism, Tautomerism and Free Energy Landscapes of a Pyridoacridinium Quaternary Salt. Marine Drugs. 2023; 21(8):446. https://doi.org/10.3390/md21080446
Chicago/Turabian StyleGartshore, Christopher J., Xiao Wang, Yongxuan Su, and Tadeusz F. Molinski. 2023. "Petrosamine Revisited. Experimental and Computational Investigation of Solvatochromism, Tautomerism and Free Energy Landscapes of a Pyridoacridinium Quaternary Salt" Marine Drugs 21, no. 8: 446. https://doi.org/10.3390/md21080446
APA StyleGartshore, C. J., Wang, X., Su, Y., & Molinski, T. F. (2023). Petrosamine Revisited. Experimental and Computational Investigation of Solvatochromism, Tautomerism and Free Energy Landscapes of a Pyridoacridinium Quaternary Salt. Marine Drugs, 21(8), 446. https://doi.org/10.3390/md21080446