
Structural Insights into the Marine Alkaloid Discorhabdin G as a Scaffold towards New Acetylcholinesterase Inhibitors
 , , , , and
, , , , and 
Abstract
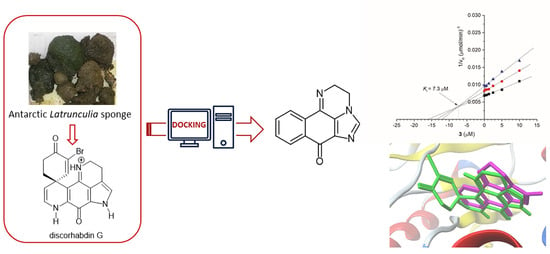
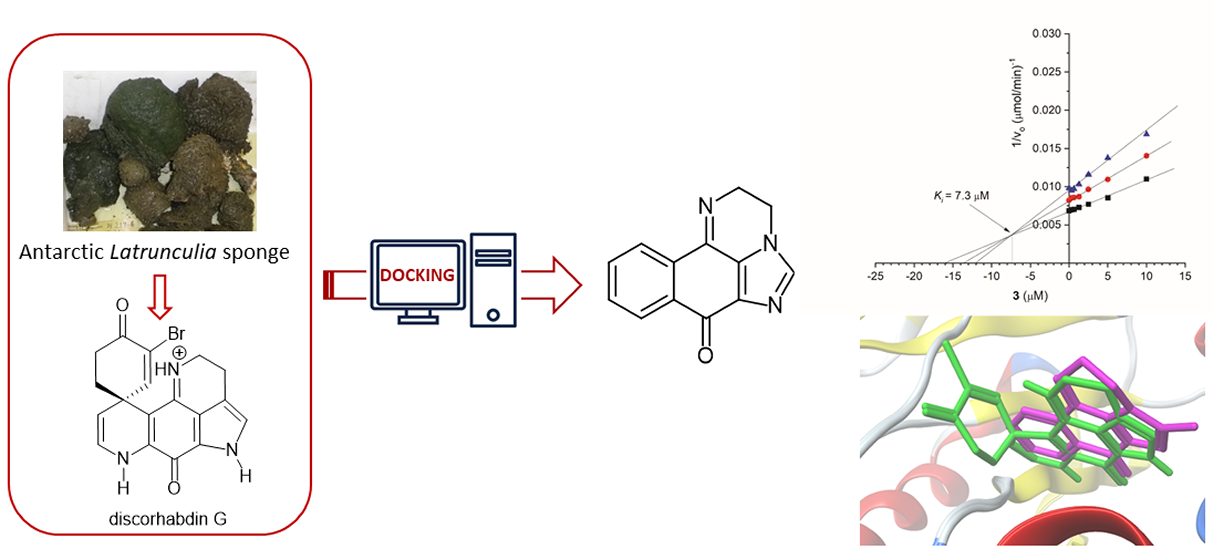
1. Introduction
2. Results and Discussion
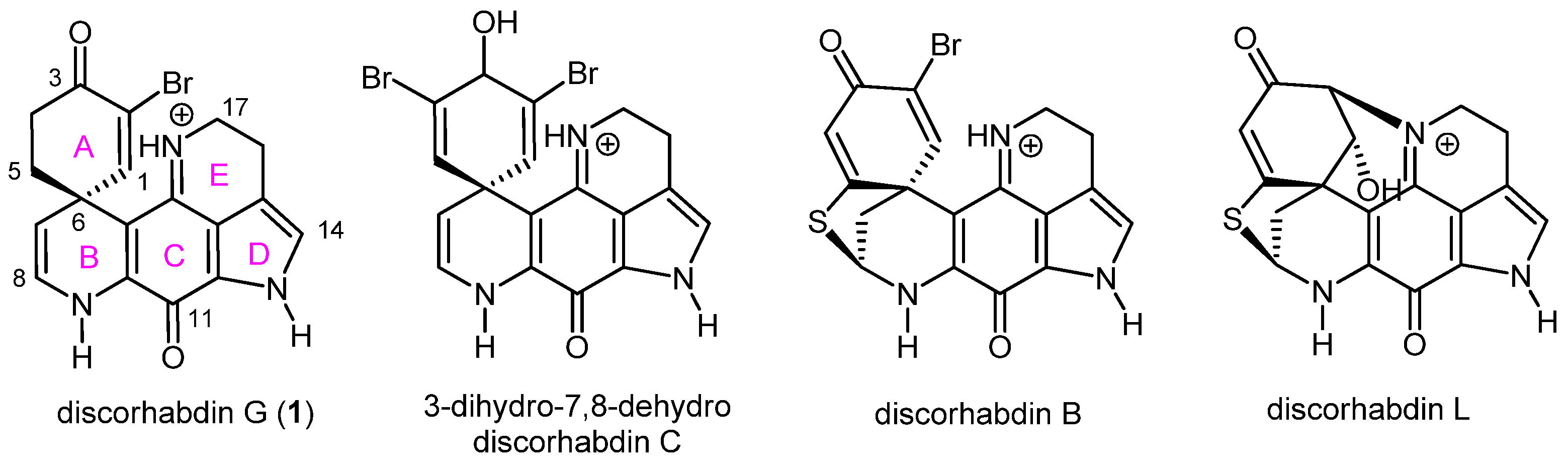
2.1. Pharmacophore Modeling of Discorhabdin G and Design of Simplified Structures
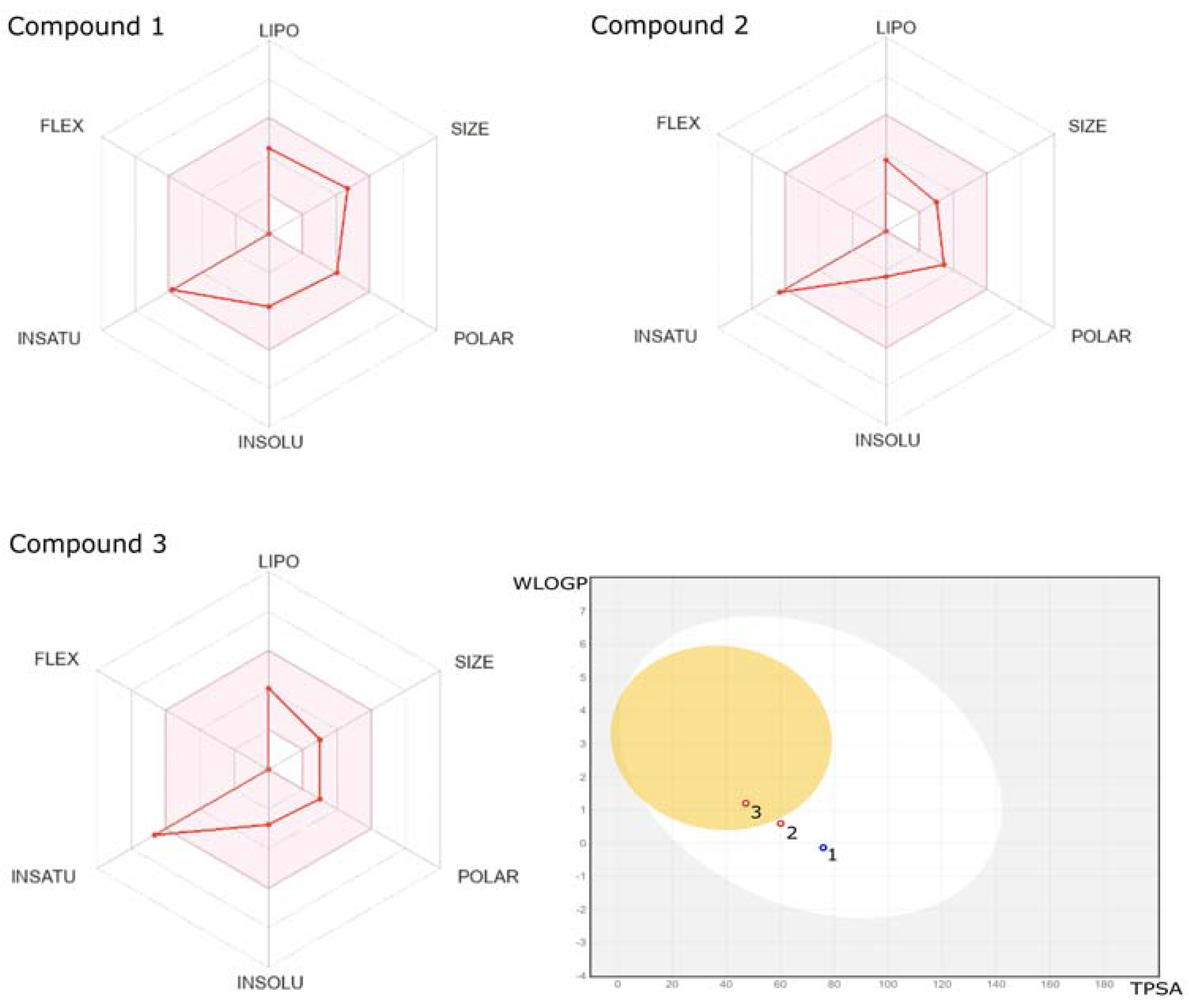
2.2. ADME/Toxicity Prediction
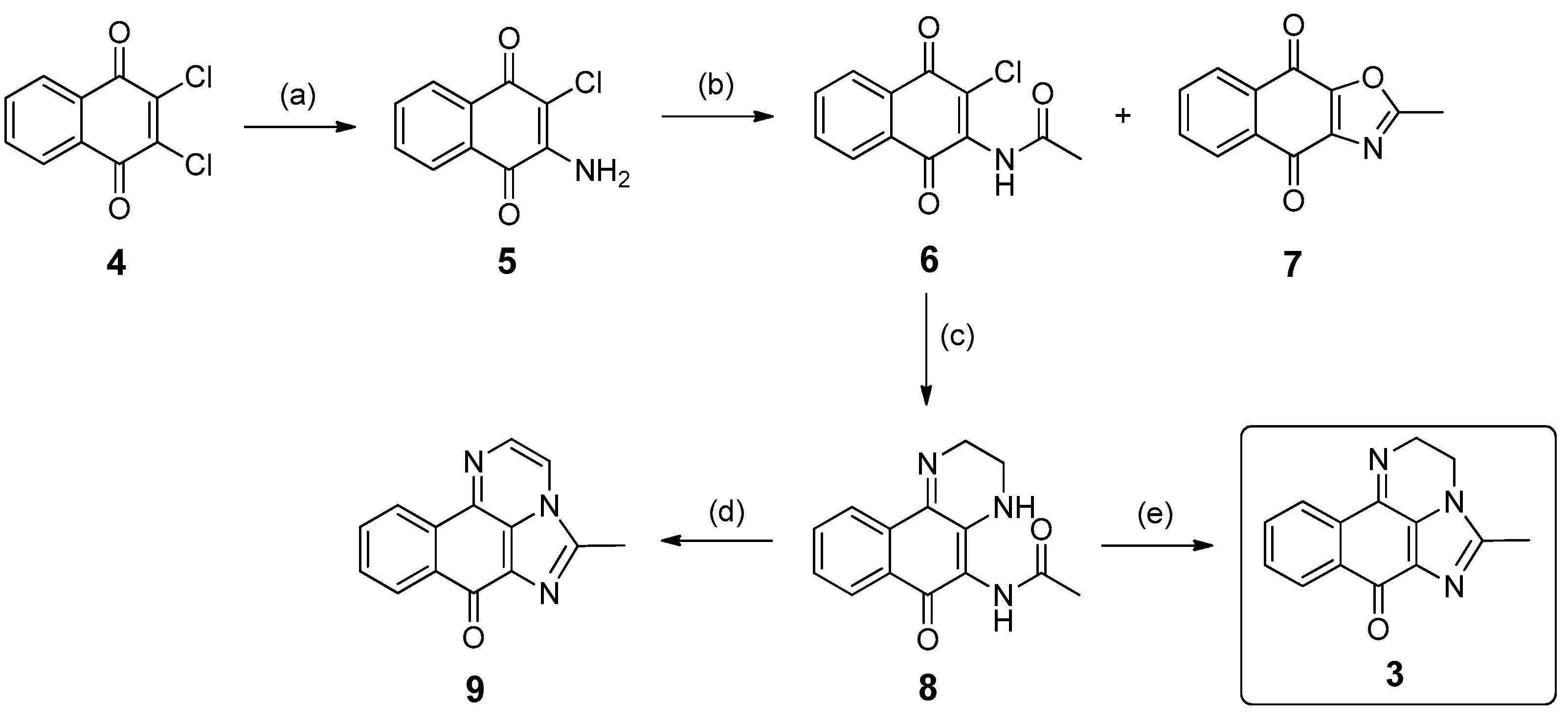
2.3. Synthesis of Compound 3
2.4. Biological Evaluation
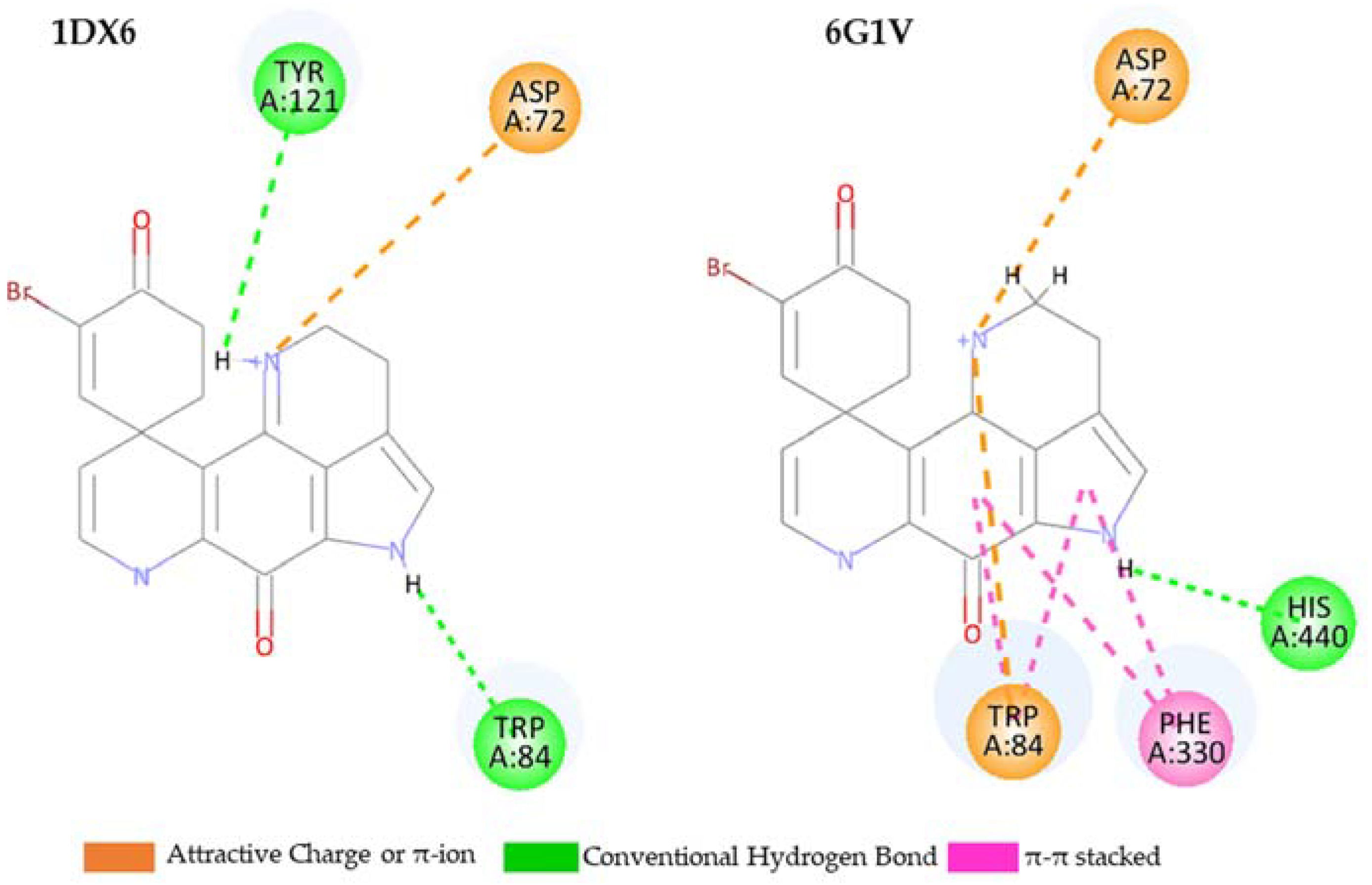
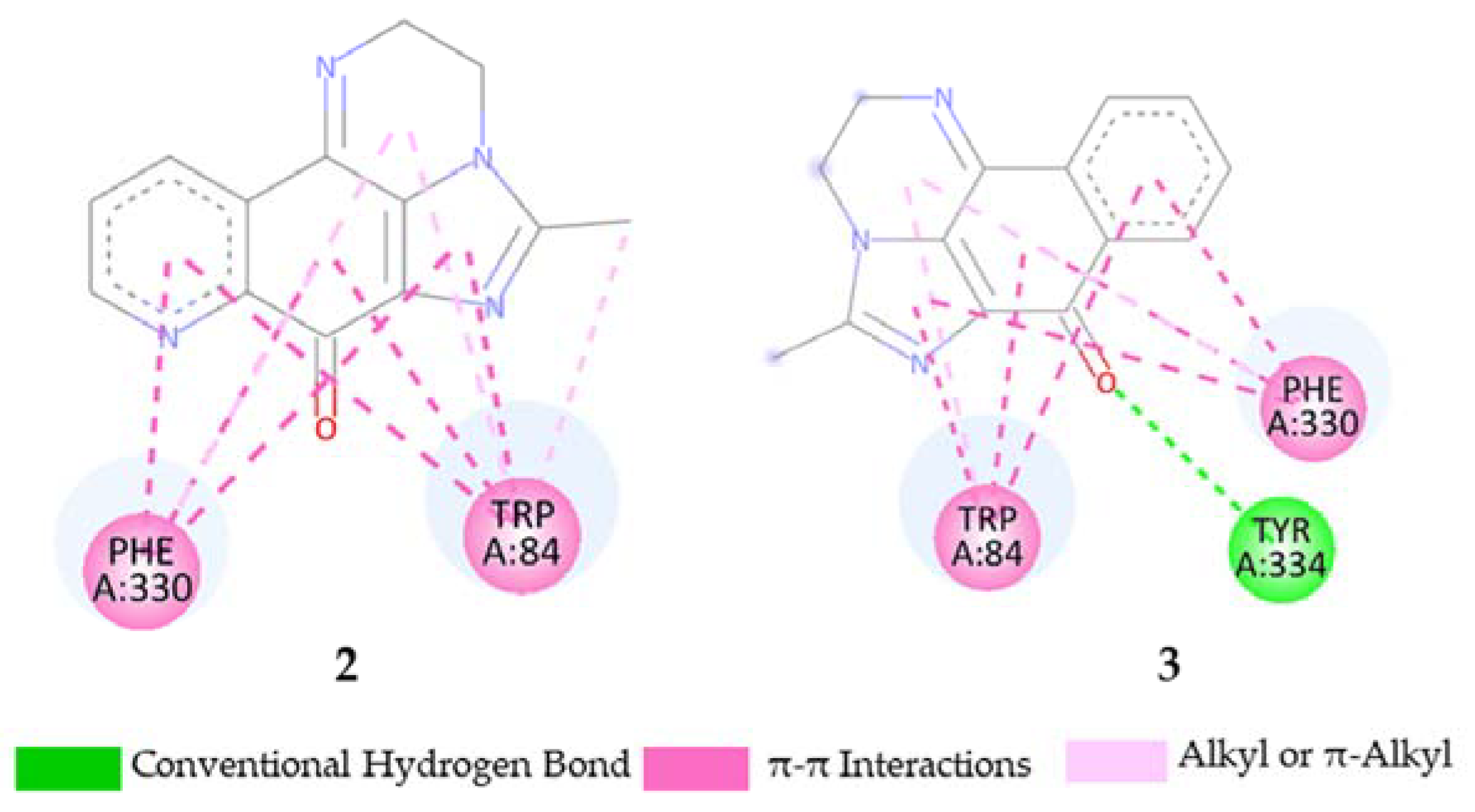
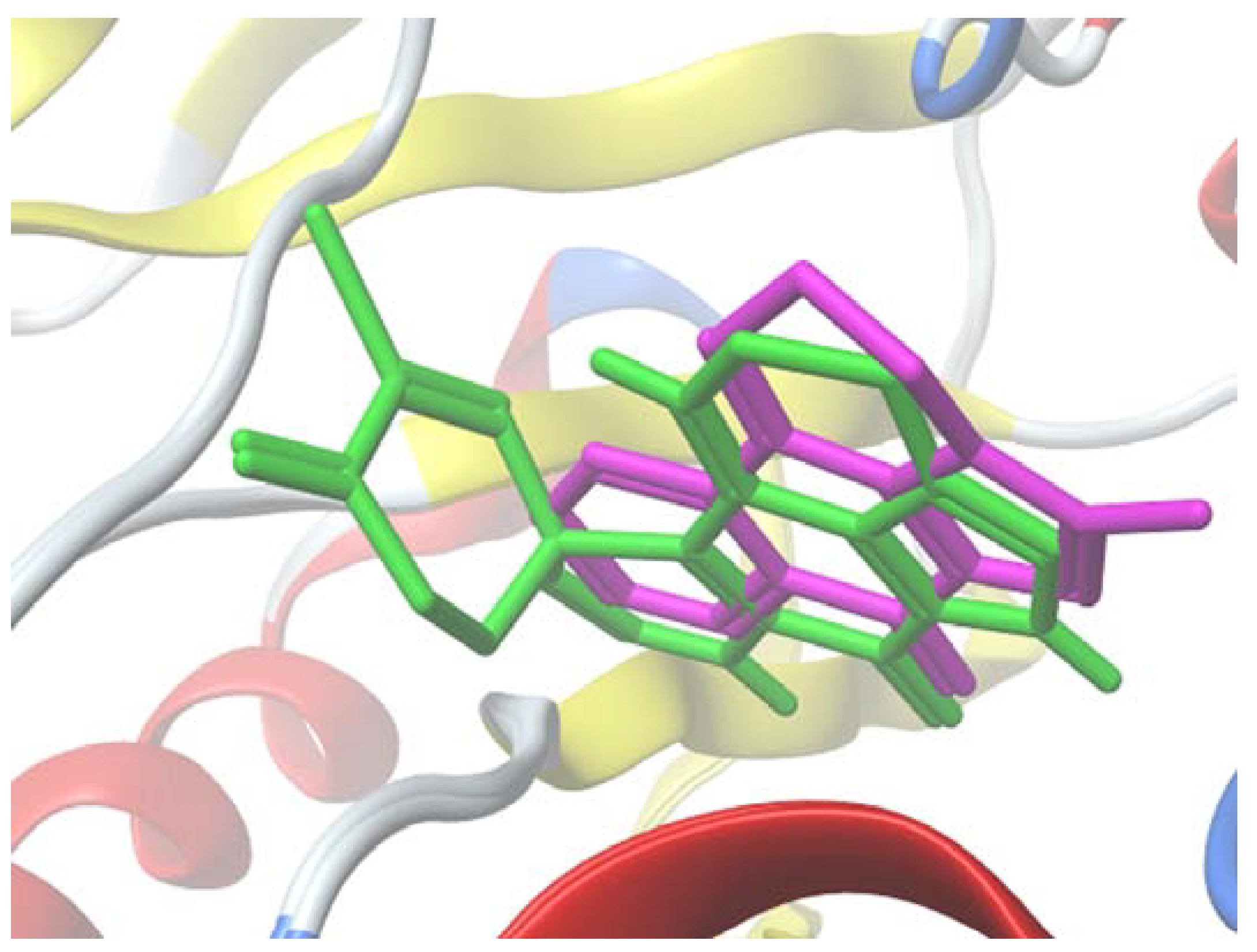
2.5. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. General Experimental Procedures
3.1.2. Synthesis of 5-Methyl-2H-benzo[h]imidazo[1,5,4-de]quinoxalin-7(3H)-one (3)
- 1.
- 2-Amino-3-chloronaphthalene-1,4-dione (5)
- 2.
- N-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)acetamide (6) and byproduct 7
- 3.
- N-(6-oxo-2,3,4,6-tetrahydrobenzo[f]quinoxalin-5-yl)acetamide (8)
- 4.
- Final product 3 and compound 9
3.2. Biological Evaluation
3.3. Computational Details
3.3.1. Prediction of Structures at Different pH Values
3.3.2. Pharmacokinetic Study
3.3.3. Docking Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perry, N.B.; Blunt, J.W.; McCombs, J.D.; Munro, M.H.G. Discorhabdin C, a highly cytotoxic pigment from a sponge of the genus Latrunculia. J. Org. Chem. 1986, 51, 5476–5478. [Google Scholar] [CrossRef]
- Hu, J.F.; Fan, H.; Xiong, J.; Wu, S.B. Discorhabdins and pyrroloiminoquinone related alkaloids. Chem. Rev. 2011, 111, 5465–5491. [Google Scholar] [CrossRef] [PubMed]
- Antunes, E.M.; Copp, B.R.; Davies-Coleman, M.T.; Samaai, T. Pyrroloiminoquinone and related metabolites from marine sponges. Nat. Prod. Rep. 2005, 22, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Fujioka, H.; Kita, Y. Synthesis of the Marine Pyrroloiminoquinone Alkaloids, Discorhabdins. Mar. Drugs 2010, 8, 1394–1416. [Google Scholar] [CrossRef] [PubMed]
- Botić, T.; Defant, A.; Zanini, P.; Žužek, M.C.; Frangež, R.; Janussen, D.; Kersken, D.; Knez, Ž.; Sepčić, K.; Mancini, I. Discorhabdin alkaloids from Antarctic Latrunculia spp. sponges as a new class of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 136, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Jin, Y.; Hou, X.; Zhu, Y.; Chen, D.; Tai, J.; Chen, Q.; Shi, C.; Ye, J.; Wu, M.; et al. Application of Marine Natural Products against Alzheimer’s Disease: Past, Present and Future. Mar. Drugs 2023, 21, 43. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.M.; Strope, J.D.; Beedie, S.L.; Huang, P.A.; Goey, A.K.L.; Cook, K.M.; Schofield, C.J.; Chau, C.H.; Cadelis, M.M.; Copp, B.R.; et al. Preclinical Evaluation of Discorhabdins in Antiangiogenic and Antitumor Models. Mar. Drugs 2018, 16, 241. [Google Scholar] [CrossRef] [PubMed]
- Orfanoudaki, M.; Emily, A.; Smith, E.A.; Hill, N.T.; Garman, K.A.; Brownell, I.; Copp, B.R.; Grkovic, T.; Henrich, C.J. An Investigation of Structure–Activity Relationships and Cell Death Mechanisms of the Marine Alkaloids Discorhabdins in Merkel Cell Carcinoma Cells. Mar. Drugs 2023, 21, 474. [Google Scholar] [CrossRef]
- Li, F.; JanusseN, D.; Tasdemir, D. New Discorhabdin B Dimers with Anticancer Activity from the Antarctic Deep-Sea Sponge Latrunculia biformis. Mar. Drugs 2020, 18, 107. [Google Scholar] [CrossRef]
- Li, F.; Pandey, P.; Janussen, D.; Chittiboyina, A.G.; Ferreira, D.; Tasdemir, D. Tridiscorhabdin and Didiscorhabdin, the First Discorhabdin Oligomers Linked with a Direct C-N Bridge from the Sponge Latrunculia biformis Collected from the Deep Sea in Antarctica. J. Nat. Prod. 2020, 83, 706–713. [Google Scholar] [CrossRef]
- Kalinski, J.-C.J.; Polyzois, A.; Waterworth, S.C.; Siwe Noundou, X.; Dorrington, R.A. Current Perspectives on Pyrroloiminoquinones: Distribution, Biosynthesis and Drug Discovery Potential. Molecules 2022, 27, 8724. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Baker, B.J.; Grimwade, J.; Alan, L.; McClintock, J.B. Discorhabdin alkakloids from the Antarctic sponge Latrunculia apicalis. J. Nat. Prod. 1995, 58, 1596–1599. [Google Scholar] [CrossRef]
- Defant, A.; Guella, G.; Mancini, I. Regioselectivity in the Multi-Component Synthesis of Indolizinoquinoline-5,12-dione Derivatives. Eur. J. Org. Chem. 2006, 2006, 4201–4210. [Google Scholar] [CrossRef]
- Defant, A.; Rossi, B.; Viliani, G.; Guella, G.; Mancini, I. Metal-assisted regioselectivity in nucleophilic substitutions: A study by Raman spectroscopy and density functional theory calculations. J. Raman Spectrosc. 2010, 41, 1398–1403. [Google Scholar] [CrossRef]
- Defant, A.; Mancini, I. Design, Synthesis and Cancer Cell Growth Inhibition Evaluation of New Aminoquinone Hybrid Molecules. Molecules 2019, 24, 2224. [Google Scholar] [CrossRef] [PubMed]
- Smyrska-Wieleba, N.; Mroczek, T. Natural Inhibitors of Cholinesterases: Chemistry, Structure-Activity and Methods of Their Analysis. Int. J. Mol. Sci. 2023, 24, 2722. [Google Scholar] [CrossRef] [PubMed]
- Truax, N.J.; Romo, D. Bridging the gap between natural product synthesis and drug discovery. Nat. Prod. Rep. 2020, 37, 1436–1453. [Google Scholar] [CrossRef] [PubMed]
- Marvinsketch. Available online: https://chemicalize.com/welcome (accessed on 25 January 2024).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and MedicinalChemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717–42728. [Google Scholar] [CrossRef]
- Swiss ADME. Available online: http://www.swissadme.ch/ (accessed on 20 December 2023).
- Molsft, L.L.C. Available online: https://www.molsoft.com/ (accessed on 15 January 2023).
- LaMarche, M.J. Think Biologically, Act Chemically. J. Med. Chem. 2022, 65, 11478–11484. [Google Scholar] [CrossRef]
- Hammam, A.S.; Osman, A.M. The Reaction of Acid Amides with 2,3-Dichloro1,4-naphthoquinone, a Novel Route to Naphth[2,3-d]oxarole-4,9-diones. J. Prakt. Chem. 1977, 319, 254–258. [Google Scholar] [CrossRef]
- Nepovimova, E.; Uliassi, E.; Korabecny, J.; Peñ Altamira, L.E.; Samez, S.; Pesaresi, A.; Garcia, G.E.; Bartolini, M.; Andrisano, V.; Bergamini, C.; et al. Multitarget Drug Design Strategy: Quinone−Tacrine Hybrids Designed to Block Amyloid-β Aggregation and To Exert Anticholinesterase and Antioxidant Effects. J. Med. Chem. 2014, 57, 8576–8589. [Google Scholar] [CrossRef] [PubMed]
- Lindon, W.K.; Moodie, L.W.K.; Sepčić, K.; Turk, T.; Frangež, R.; Svenson, J. Natural cholinesterase inhibitors from marine organisms. Nat. Prod. Rep. 2019, 36, 1053–1092. [Google Scholar] [CrossRef]
- Soukup, O.; Winder, M.; Killi, U.K.; Wsol, V.; Jun, D.; Kuca, K.; Tobin, G. Acetylcholinesterase Inhibitors and Drugs Acting on Muscarinic Receptors-Potential Crosstalk of Cholinergic Mechanisms During Pharmacological Treatment. Curr. Neuropharmacol. 2017, 15, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, H.; Chen, Y.; Sun, H. Recent progress in the identification of the selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity, Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. 4. A new dynamical correlation functional and implications for exactexchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- O’Boyle, N.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. jCompoundMapper: An open source Java library and command-line tool for chemical fingerprints. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Mod. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Galdeano, C.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Bartolini, M.; Perez, B.; Clos, M.V.; Silman, I.; Jean, L.; Colletier, J.P.; Renard, P.Y.; et al. Increasing Polarity in Tacrine and Huprine Derivatives: Potent Anticholinesterase Agents for the Treatment of Myasthenia Gravis. Molecules 2018, 23, 634. [Google Scholar] [CrossRef]
- Greenblatt, H.M.; Kryger, G.; Lewis, T.T.; Silman, I.; Sussman, J.L. Structure of Acetylcholinesterase Complexed with (-)-Galanthamine at 2. 3A Resolution. FEBS Lett. 1999, 463, 321–326. [Google Scholar] [CrossRef]
- Cheung, J.; Gary, E.N.; Shiomi, K.; Rosenberry, T.L. Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility. Med. Chem. Lett. 2013, 4, 1091e1096. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design; Lecture Notes in Computer Science 4150; Springer: Berlin/Heidelberg, Germany, 2006; pp. 247–258. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA Discovery Studio Visualizer 21.1.0.0 (Discovery Studio 2021 Client, Dassault Systèmes). Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 25 March 2024).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Number of H-bond acceptors | 2.00 | 4.00 | 3.00 |
| Number of H-bond donors | 3.00 | 0.00 | 0.00 |
| Consensus Log P | 1.78 | 0.97 | 1.68 |
| WLOGP | −0.13 | 0.60 | 1.21 |
| TPSA (in Å2) | 75.93 | 60.14 | 47.25 |
| BBB | No | No | Yes |
| BBB 1 | 3.99 | 4.55 | 4.96 |
| Gastrointestinal (GI) absorption | High | High | High |
| Compound | eeAChE | hAChE | BChE | |||
|---|---|---|---|---|---|---|
| IC50 (µM) | Ki (µM) | IC50 (µM) | Ki (µM) | IC50 (µM) | Ki (µM) | |
| Discorhabdin G (1) a | 1.3 | 1.6 | 116 | 56.2 | 7.0 | 5.0 |
| Physostigmine salicylate a | 3.0 | 14.5 | 28.5 | |||
| 3 | 13.5 | 7.3 | 16.9 | 5.8 | >400 | n.d. |
| 7 | 187.6 | 65 | 234.5 | 50 | 145.4 | 50 |
| 8 | >400 | n.d. | >400 | n.d. | >400 | n.d. |
| Neostigmine methylsulfate | 6.0 | 7.5 | 83.7 | |||
| Physostigmine salicylate | 7.3 | 7.3 | 14.5 | |||
| Torpedo californica AChE (6G1V) | Torpedo californica AChE (1DX6) | Homo sapiens AChE (4M0E) | ||||
|---|---|---|---|---|---|---|
| Compound | E (Vina) kcal/mol | PLANTS Score | E(Vina) kcal/mol | Score PLANTS | E(Vina) kcal/mol | Score PLANTS |
| 1 | −12.133 | −102.951 | −11.300 | −82.6079 | −9.990 | −89.4937 |
| 3 | −9.483 | −94.6408 | −9.652 | −83.204 | −9.374 | −89.3812 |
| 2 | −8.620 | −90.7618 | −9.247 | −82.404 | −8.541 | −87.5673 |
| 7 | −8.573 | −82.8096 | −8.798 | −74.2019 | −8.771 | −83.251 |
| physostigmine | −9.291 | −86.9757 | −9.652 | −93.3553 | −7.981 | −85.8552 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Defant, A.; Carloni, G.; Innocenti, N.; Trobec, T.; Frangež, R.; Sepčić, K.; Mancini, I. Structural Insights into the Marine Alkaloid Discorhabdin G as a Scaffold towards New Acetylcholinesterase Inhibitors. Mar. Drugs 2024, 22, 173. https://doi.org/10.3390/md22040173
Defant A, Carloni G, Innocenti N, Trobec T, Frangež R, Sepčić K, Mancini I. Structural Insights into the Marine Alkaloid Discorhabdin G as a Scaffold towards New Acetylcholinesterase Inhibitors. Marine Drugs. 2024; 22(4):173. https://doi.org/10.3390/md22040173
Chicago/Turabian StyleDefant, Andrea, Giacomo Carloni, Nicole Innocenti, Tomaž Trobec, Robert Frangež, Kristina Sepčić, and Ines Mancini. 2024. "Structural Insights into the Marine Alkaloid Discorhabdin G as a Scaffold towards New Acetylcholinesterase Inhibitors" Marine Drugs 22, no. 4: 173. https://doi.org/10.3390/md22040173
APA StyleDefant, A., Carloni, G., Innocenti, N., Trobec, T., Frangež, R., Sepčić, K., & Mancini, I. (2024). Structural Insights into the Marine Alkaloid Discorhabdin G as a Scaffold towards New Acetylcholinesterase Inhibitors. Marine Drugs, 22(4), 173. https://doi.org/10.3390/md22040173