
Genomic Analysis of Novel Sulfitobacter Bacterial Strains Isolated from Marine Biofilms


Abstract
:1. Introduction
2. Results
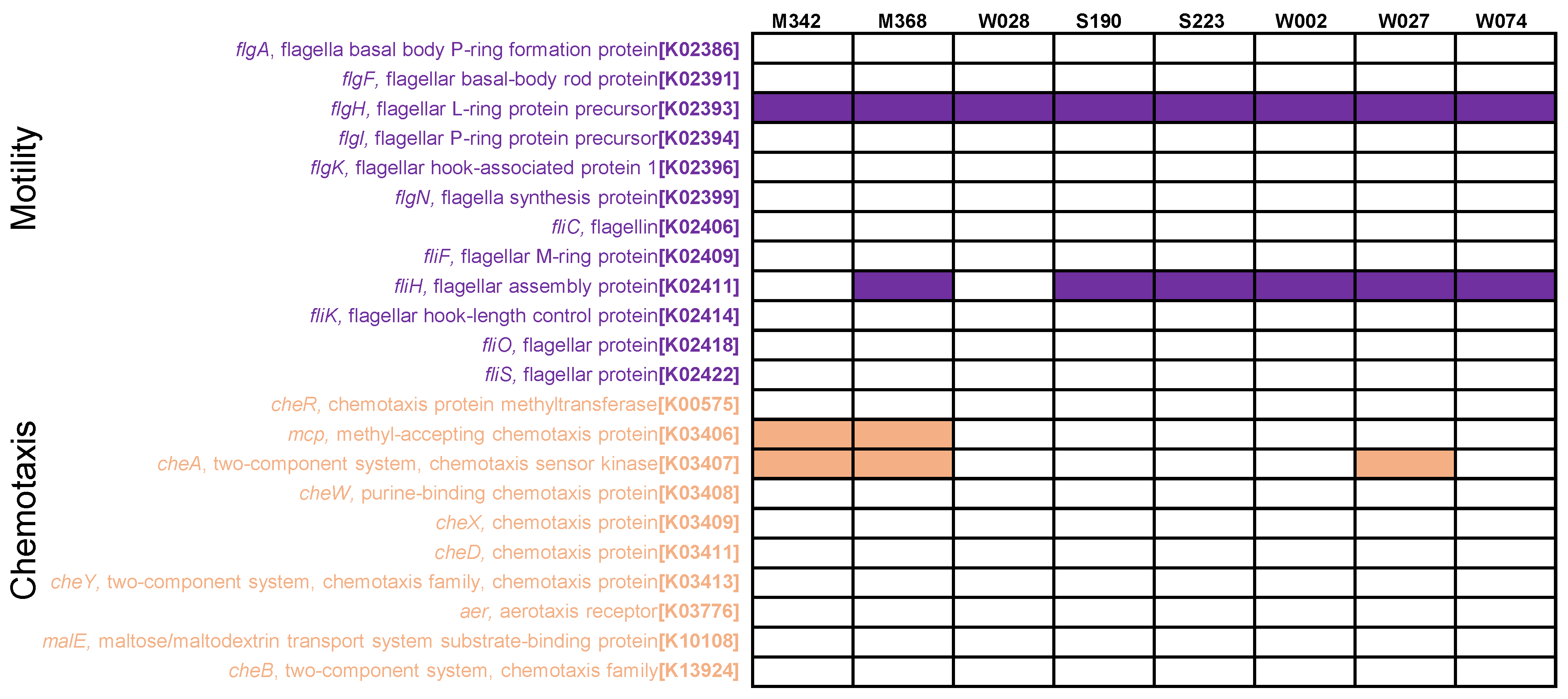
2.1. Phenotype and Phylogeny
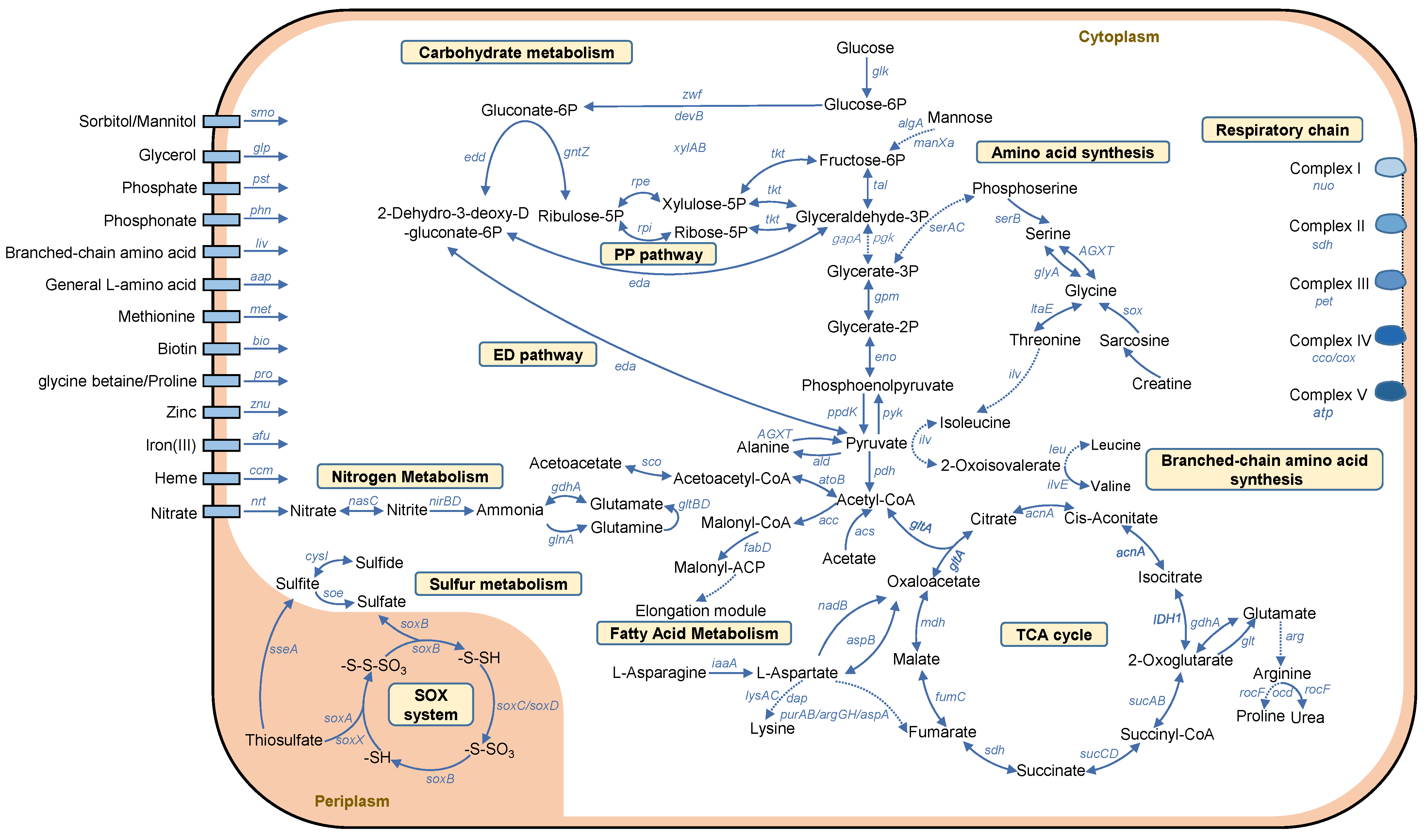
2.2. Metabolic Pathways
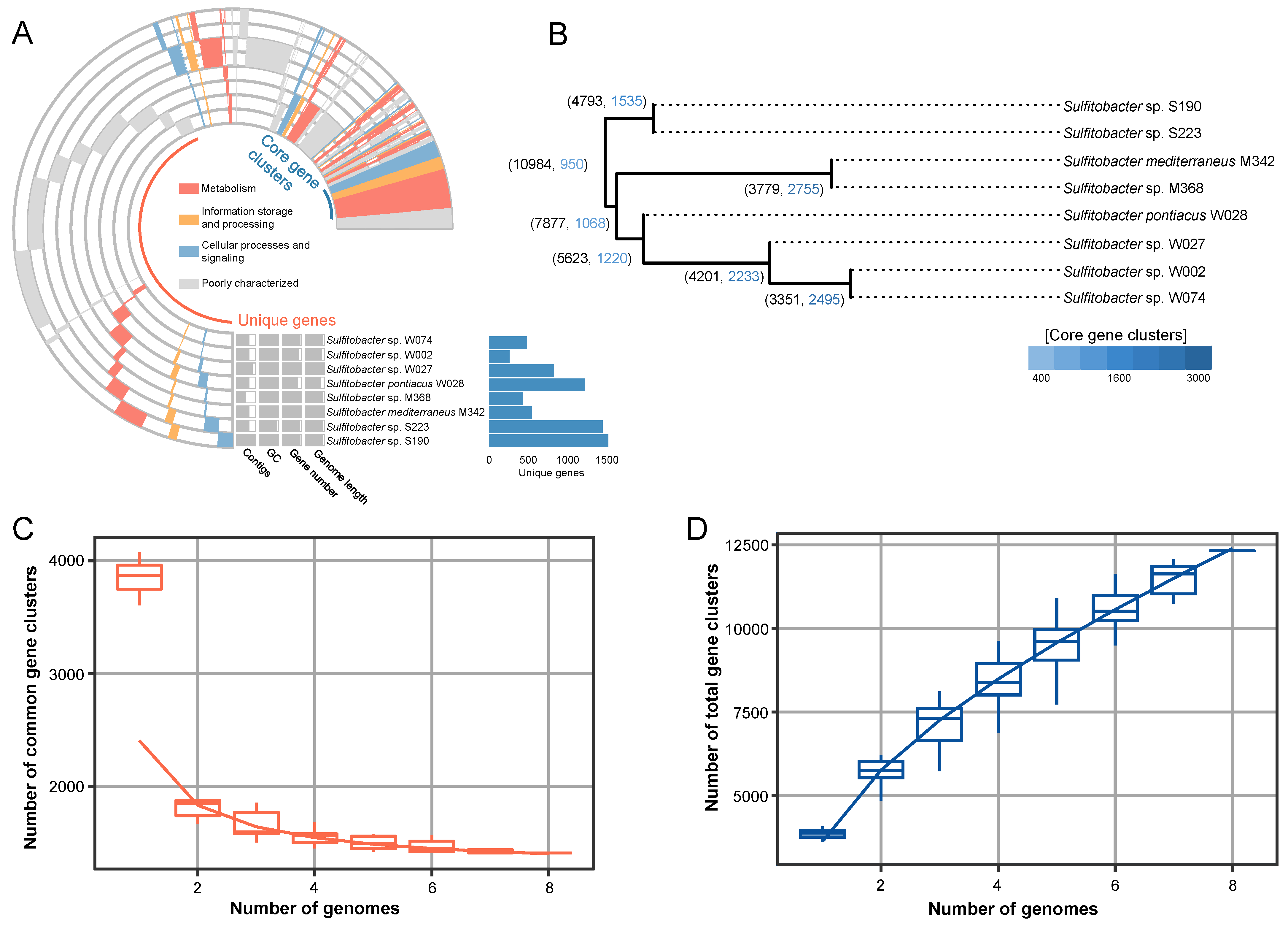
2.3. Pangenome Analyses
2.4. Potential of Strain for Biosynthesis of Secondary Metabolites
2.5. In Situ Biosynthetic Gene Expression
3. Discussion
4. Experimental Procedures
4.1. Biofilm Sampling and Strain Isolation
4.2. Observation of Bacterial Morphology with TEM
4.3. Genomic Sequencing and Assembly
4.4. Basic Genomic Analyses
4.5. Phylogenetic Tree Building
4.6. Metabolic Pathway Reconstruction and Pangenome Analysis
4.7. Analysis of Biosynthetic Potential of Strains
4.8. Metatranscriptomic Analysis
4.9. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sorokin, D.Y. Sulfitobacter pontiacus gen. nov., sp. nov.—A new heterotrophic bacterium from the Black Sea, specialized on sulfite oxidation. Microbiology 1995, 64, 354–365. [Google Scholar]
- Curson, A.R.J.; Rogers, R.; Todd, J.D.; Brearley, C.A.; Johnston, A.W.B. Molecular genetic analysis of a dimethylsulfoniopropionate lyase that liberates the climate-changing gas dimethylsulfide in several marine alpha-proteobacteria and Rhodobacter sphaeroides. Environ. Microbiol. 2008, 10, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Pukall, R.; Buntefuss, D.; Fruhling, A.; Rohde, M.; Kroppenstedt, R.M.; Burghardt, J.; Lebaron, P.; Bernard, L.; Stackebrandt, E. Sulfitobacter mediterraneus sp. nov., a new sulfite-oxidizing member of the alpha-proteobacteria. Int. J. Syst. Bacteriol. 1999, 49, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Amberkar, U.; Khandeparker, R.; D Menezes, L.; Meena, R.M. Phylogenetic diversity of culturable marine bacteria from sediments underlying the oxygen minimum zone of the Arabian Sea and their role in nitrate reduction. Mar. Ecol. 2021, 42, e12646. [Google Scholar] [CrossRef]
- Petersen, J.; Brinkmann, H.; Bunk, B.; Michael, V.; Päuker, O.; Pradella, S. Think pink: Photosynthesis, plasmids and the Roseobacter clade. Environ. Microbiol. 2012, 14, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Guo, Y.; Huang, Z.; Tang, K.; Wang, X. Comparative genomic analysis of cold-water coral-derived Sulfitobacter faviae: Insights into their habitat adaptation and metabolism. Mar. Drugs 2023, 21, 309. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Ge, Y.M.; Iqbal, N.M.; Yang, X.; Zhang, X.L. Sulfitobacter alexandrii sp. nov., a new microalgae growth-promoting bacterium with exopolysaccharides bioflocculanting potential isolated from marine phycosphere. Antonie Van Leeuwenhoek 2021, 114, 1091–1106. [Google Scholar] [PubMed]
- Beiralas, R.; Ozer, N.; Segev, E. Abundant Sulfitobacter marine bacteria protect Emiliania huxleyi algae from pathogenic bacteria. ISME Commun. 2023, 3, 100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ding, W.; Yang, B.; Tian, R.; Gu, S.; Luo, H.; Qian, P.Y. Genomic and transcriptomic evidence for carbohydrate consumption among microorganisms in a cold seep brine pool. Front. Microbiol. 2016, 7, 1825. [Google Scholar] [CrossRef]
- Wei, Z.; Zhao, L.; Wang, S.; Chang, L.; Shi, J.; Kong, X.; Li, M.; Lin, J.; Zhang, W.; Bao, Z.; et al. Paralytic shellfish toxins producing dinoflagellates cause dysbacteriosis in scallop gut microbial biofilms. Ecotox. Environ. Safe. 2024, 273, 116146. [Google Scholar] [CrossRef]
- Qin, P.; Cui, H.; Li, P.; Wang, S.; Fan, S.; Lu, J.; Sun, M.; Zhang, H.; Wang, S.; Su, X.; et al. Early stage of biofilm assembly on microplastics is structured by substrate size and bacterial motility. iMeta 2023, 2, e121. [Google Scholar] [CrossRef] [PubMed]
- Qian, P.Y.; Cheng, A.; Wang, R.; Zhang, R. Marine biofilms: Diversity, interactions and biofouling. Nat. Rev. Microbiol. 2022, 20, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.C.; Wuertz, S. Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol. 2019, 17, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ding, W.; Li, Y.X.; Tam, C.; Bougouffa, S.; Wang, R.; Pei, B.; Chiang, H.; Leung, P.; Lu, Y.; et al. Marine biofilms constitute a bank of hidden microbial diversity and functional potential. Nat. Commun. 2019, 10, 517. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Wang, S.; Qin, P.; Fan, S.; Su, X.; Cai, P.; Lu, J.; Cui, H.; Wang, M.; Shu, Y.; et al. Anaerobic thiosulfate oxidation by the Roseobacter group is prevalent in marine biofilms. Nat. Commun. 2023, 14, 2033. [Google Scholar] [CrossRef] [PubMed]
- Michael, V.; Frank, O.; Bartling, P.; Scheuner, C.; Göker, M.; Brinkmann, H.; Petersen, J. Biofilm plasmids with a rhamnose operon are widely distributed determinants of the ‘swim-or-stick’lifestyle in roseobacters. ISME J. 2016, 10, 2498–2513. [Google Scholar] [CrossRef] [PubMed]
- Fei, C.; Ochsenkühn, M.A.; Shibl, A.A.; Isaac, A.; Wang, C.; Amin, S.A. Quorum sensing regulates ‘swim-or-stick’ lifestyle in the phycosphere. Environ. Microbiol. 2020, 22, 4761–4778. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lu, J.; Qin, P.; Wang, S.; Ding, W.; Fu, H.H.; Zhang, Y.Z.; Zhang, W. Biofilm formation stabilizes metabolism in a Roseobacteraceae bacterium under temperature increase. Appl. Environ. Microbiol. 2023, 89, e00601–e00623. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Zeng, J.; Wang, X.; Drlica, K.; Zhao, X. Post-stress bacterial cell death mediated by reactive oxygen species. Proc. Natl Acad. Sci. USA 2019, 116, 10064–10071. [Google Scholar] [CrossRef]
- Flamholz, A.; Noor, E.; Bar-Even, A.; Liebermeister, W.; Milo, R. Glycolytic strategy as a tradeoff between energy yield and protein cost. Proc. Natl. Acad. Sci. USA 2013, 110, 10039–10044. [Google Scholar] [CrossRef]
- Czajka, J.J.; Abernathy, M.H.; Benites, V.T.; Baidoo, E.E.K.; Deming, J.W.; Tang, Y.J. Model metabolic strategy for heterotrophic bacteria in the cold ocean based on Colwellia psychrerythraea 34H. Proc. Natl. Acad. Sci. USA 2018, 115, 12507–12512. [Google Scholar] [CrossRef]
- Chavarría, M.; Nikel, P.I.; Pérez-Pantoja, D.; de Lorenzo, V. The entner-doudoroff pathway empowers Pseudomonas putida KT2440 with a high tolerance to oxidative stress. Environ. Microbiol. 2013, 15, 1772–1785. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Alam, M.T.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Klingner, A.; Bartsch, A.; Dogs, M.; Wagner-Dobler, I.; Jahn, D.; Simon, M.; Brinkhoff, T.; Becker, J.; Wittmann, C. Large-Scale 13C flux profiling reveals conservation of the Entner-Doudoroff pathway as a glycolytic strategy among marine bacteria that use glucose. Appl. Environ. Microbiol. 2015, 81, 2408–2422. [Google Scholar] [CrossRef] [PubMed]
- Hobmeier, K.; Goëss, M.C.; Sehr, C.; Schwaminger, S.; Berensmeier, S.; Kremling, A.; Kunte, H.J.; Pflüger-Grau, K.; Marin-Sanguino, A. Anaplerotic pathways in Halomonas elongata: The role of the sodium gradient. Front. Microbiol. 2020, 11, 561800. [Google Scholar] [CrossRef] [PubMed]
- Mullis, M.M.; Selwyn, J.D.; Kevorkian, R.; Tague, E.D.; Castro, H.F.; Campagna, S.R.; Lloyd, K.G.; Reese, B.K. Microbial survival mechanisms within serpentinizing Mariana forearc sediments. FEMS Microbiol. Ecol. 2023, 99, fiad003. [Google Scholar] [CrossRef]
- Su, X.; Cui, H.; Zhang, W. Copiotrophy in a marine-biofilm-derived Roseobacteraceae bacterium can be supported by amino acid metabolism and thiosulfate oxidation. Int. J. Mol. Sci. 2023, 24, 8617. [Google Scholar] [CrossRef]
- Paoli, L.; Ruscheweyh, H.J.; Forneris, C.C.; Hubrich, F.; Kautsar, S.; Bhushan, A.; Lotti, A.; Clayssen, Q.; Salazar, G.; Milanese, A.; et al. Biosynthetic potential of the global ocean microbiome. Nature 2022, 607, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Hu, G.A.; Zhou, Z.Y.; Yu, W.C.; Du, A.Q.; Yang, C.L.; Yu, Y.L.; Chen, J.W.; Zhang, H.W.; Wu, Q.; et al. Global analysis of the biosynthetic chemical space of marine prokaryotes. Microbiome 2023, 11, 144. [Google Scholar] [CrossRef]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef]
- Ayikpoe, R.S.; Shi, C.; Battiste, A.J.; Eslami, S.M.; Ramesh, S.; Simon, M.A.; Bothwell, I.R.; Lee, H.; Rice, A.J.; Ren, H.; et al. A scalable platform to discover antimicrobials of ribosomal origin. Nat. Commun. 2022, 13, 6135. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.H.; Vior, N.M.; Hems, E.S.; Lacret, R.; Truman, A.W. Discovery and characterisation of an amidine-containing ribosomally-synthesised peptide that is widely distributed in nature. Chem. Sci. 2021, 12, 11769–11778. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, S.; Ma, S.; Deng, Z.; Ding, W.; Zhang, Q. Radical SAM-dependent ether crosslink in daropeptide biosynthesis. Nat. Commun. 2022, 13, 2361. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; He, B.B.; Lei, K.; Gao, Y.; Shi, Y.; Zhong, Z.; Liu, H.; Liu, R.; Zhang, H.; Wu, S.; et al. Rule-based omics mining reveals antimicrobial macrocyclic peptides against drug-resistant clinical isolates. Nat. Commun. 2024, 15, 4901. [Google Scholar] [CrossRef] [PubMed]
- Ayikpoe, R.S.; Zhu, L.; Chen, J.Y.; Ting, C.P.; Van Der Donk, W.A. Macrocyclization and backbone rearrangement during RiPP biosynthesis by a SAM-dependent domain-of-unknown-function 692. ACS Cent. Sci. 2023, 9, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- He, B.B.; Liu, J.; Cheng, Z.; Liu, R.; Zhong, Z.; Gao, Y.; Liu, H.; Song, Z.M.; Tian, Y.; Li, Y.X. Bacterial Cytochrome P450 Catalyzed Post-translational Macrocyclization of Ribosomal Peptides. Angew. Chem. Int. Ed. 2023, 62, e202311533. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, Z.; Kuipers, O.P. Mimicry of a non-ribosomally produced antimicrobial, brevicidine, by ribosomal synthesis and post-translational modification. Cell Chem. Biol. 2020, 27, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kuipers, O.P. Nisin-and ripcin-derived hybrid lanthipeptides display selective antimicrobial activity against Staphylococcus aureus. ACS Synth. Biol. 2021, 10, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- González-Torres, P.; Gabaldón, T. Genome variation in the model halophilic bacterium Salinibacter ruber. Front. Microbiol. 2018, 9, 372511. [Google Scholar] [CrossRef]
- Simonsen, A.K. Environmental stress leads to genome streamlining in a widely distributed species of soil bacteria. ISME J. 2022, 16, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Wu, M.; Scott, A.J. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 2012, 28, 1033–1034. [Google Scholar] [CrossRef]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, Y.; Fan, G.; Sun, D.; Zhang, X.; Yu, Z.; Wang, J.; Wu, L.; Shi, W.; Ma, J. IPGA: A handy integrated prokaryotes genome and pan-genome analysis web service. iMeta 2022, 1, e55. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; Van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; van der Hooft, J.J.; de Ridder, D.; Medema, M.H. BiG-SLiCE: A highly scalable tool maps the diversity of 1.2 million biosynthetic gene clusters. Gigascience 2021, 10, giaa154. [Google Scholar] [CrossRef] [PubMed]
- Terlouw, B.R.; Blin, K.; Navarro-Muñoz, J.C.; Avalon, N.E.; Chevrette, M.G.; Egbert, S.; Lee, S.; Meijer, D.; Recchia, M.J.J.; Reitz, Z.L.; et al. MIBiG 3.0: A community-driven effort to annotate experimentally validated biosynthetic gene clusters. Nucleic Acids Res. 2023, 51, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Contigs | Chromosomes | Plasmids | Total Length (bp) | GC Content (%) | CDS | rRNA | tRNA | ORFs | KEGG-Annotated ORFs | NCBI Accession Number |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M368 | 3 | 1 | 2 | 4,014,743 | 58.52 | 3944 | 6 | 44 | 3947 | 2942 | CP081113-CP081115 |
| W027 | 5 | 1 | 4 | 4,147,109 | 60.24 | 4013 | 9 | 52 | 4022 | 3001 | CP083564-CP083568 |
| W002 | 4 | 1 | 3 | 3,707,314 | 61.42 | 3605 | 12 | 51 | 3611 | 2691 | CP081126-CP081129 |
| M342 | 4 | 1 | 3 | 4,223,094 | 58.01 | 4073 | 6 | 46 | 4077 | 3046 | CP081109- CP081112 |
| W028 | 4 | 1 | 3 | 3,499,419 | 60.45 | 3375 | 9 | 49 | 3380 | 2553 | CP081116-CP081119 |
| S190 | 6 | 1 | 5 | 3,966,771 | 61.48 | 3867 | 6 | 47 | 3872 | 2836 | CP081120-CP081125 |
| S223 | 4 | 1 | 3 | 4,030,883 | 56.25 | 3800 | 6 | 44 | 3802 | 2773 | CP083560-CP083563 |
| W074 | 4 | 1 | 3 | 3,976,848 | 60.96 | 3880 | 12 | 52 | 3888 | 2872 | CP081130-CP081133 |
| Sample Name | Sampling Time | NCBI Accession Number | Clean Reads | Read Length (bp) | Contigs | Average Length of Contigs (bp) | ORFs |
|---|---|---|---|---|---|---|---|
| Metatranscriptome-1 | September 2020 | SAMN21619182 | 220,546,791 × 2 | 150 | 2,515,854 | 510.4 | 2,999,230 |
| Metatranscriptome-2 | November 2020 | SAMN21619183 | 230,543,188 × 2 | 150 | 3,111,162 | 498.8 | 3,703,787 |
| Metatranscriptome-3 | January 2021 | SAMN21619184 | 241,275,710 × 2 | 150 | 3,316,686 | 476 | 1,242,031 |
| Metatranscriptome-4 | March 2021 | SAMN21619185 | 208,807,982 × 2 | 150 | 3,845,996 | 513.2 | 1,537,969 |
| Metatranscriptome-5 | May 2021 | PRJNA753157 | 218,063,438 × 2 | 150 | 855,508 | 626.1 | 402,727 |
| Metatranscriptome-6 | July 2021 | PRJNA753157 | 272,768,371 × 2 | 150 | 1,124,063 | 310.9 | 1,189,877 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, H.; Fan, S.; Ding, W.; Zhang, W. Genomic Analysis of Novel Sulfitobacter Bacterial Strains Isolated from Marine Biofilms. Mar. Drugs 2024, 22, 289. https://doi.org/10.3390/md22070289
Cui H, Fan S, Ding W, Zhang W. Genomic Analysis of Novel Sulfitobacter Bacterial Strains Isolated from Marine Biofilms. Marine Drugs. 2024; 22(7):289. https://doi.org/10.3390/md22070289
Chicago/Turabian StyleCui, Han, Shen Fan, Wei Ding, and Weipeng Zhang. 2024. "Genomic Analysis of Novel Sulfitobacter Bacterial Strains Isolated from Marine Biofilms" Marine Drugs 22, no. 7: 289. https://doi.org/10.3390/md22070289