
Molecular Dynamics Simulation Reveal the Structure–Activity Relationships of Kainoid Synthases


Abstract
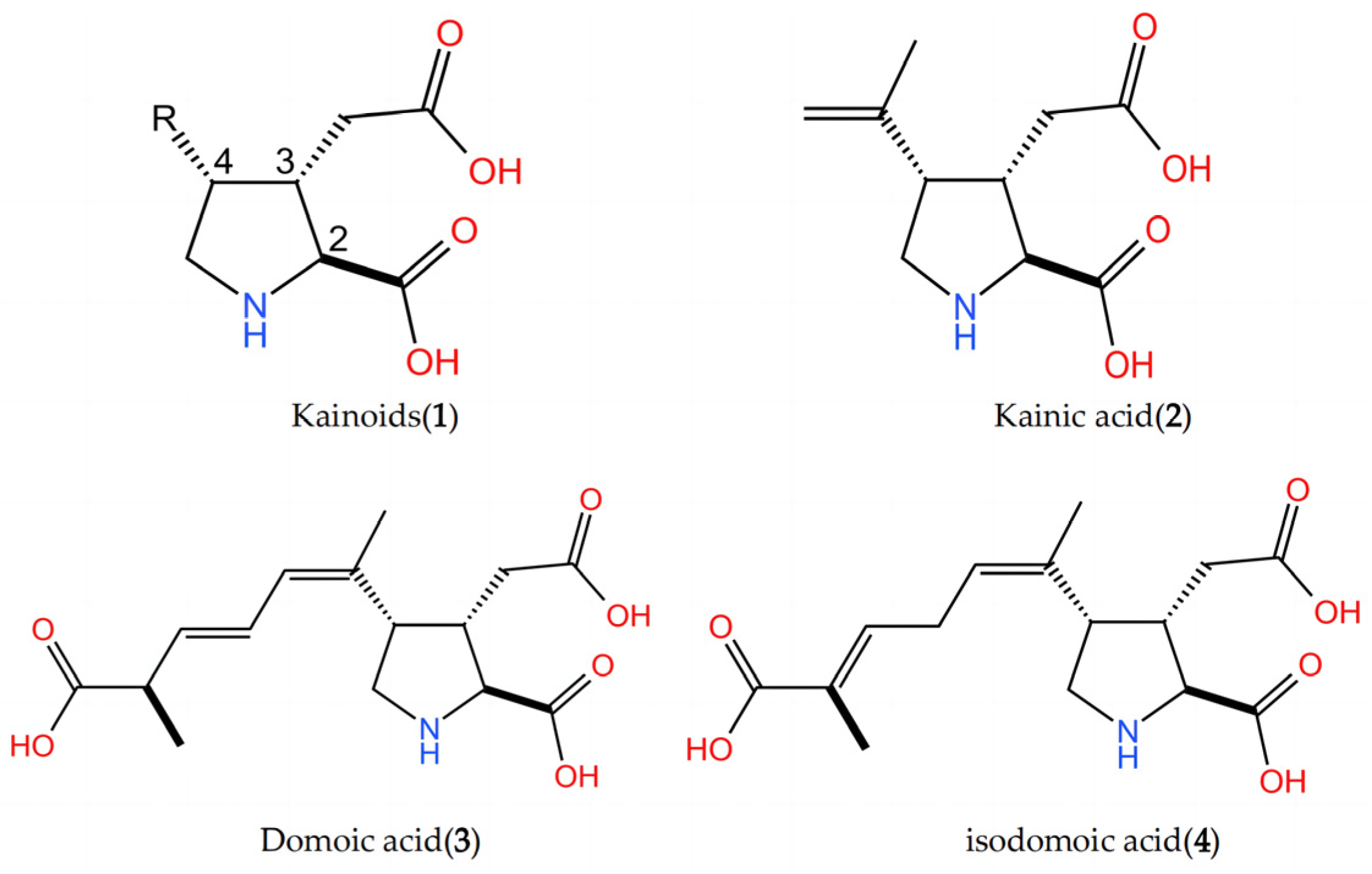
1. Introduction
2. Results and Discussion
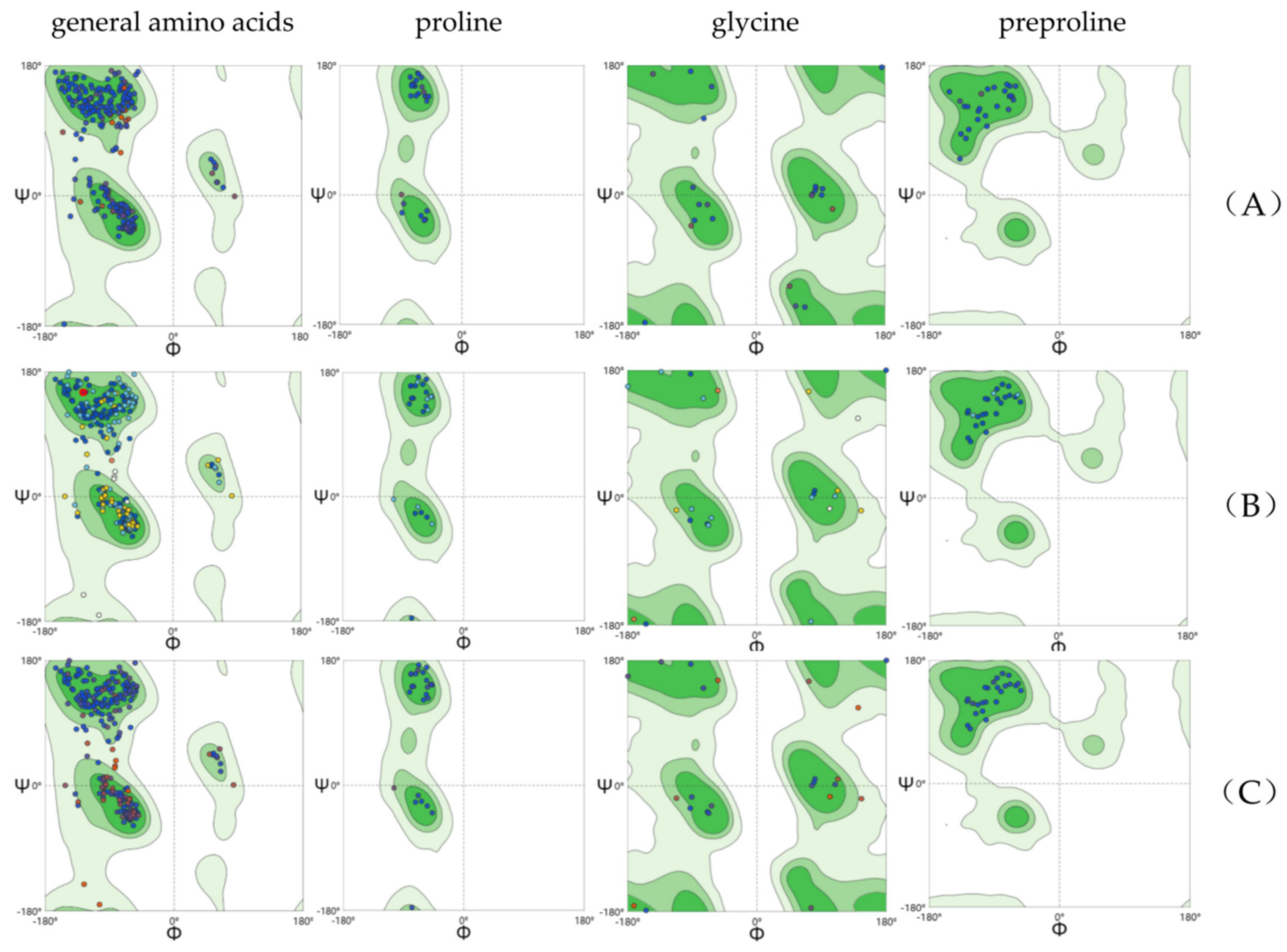
2.1. AlphaFold2 Predicts the Kainoid Synthases Structure
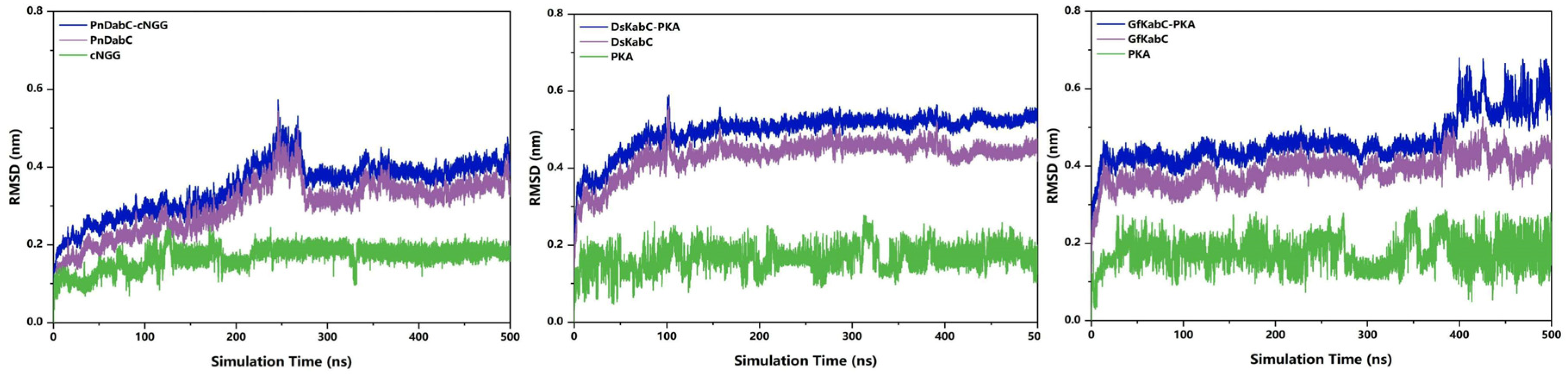
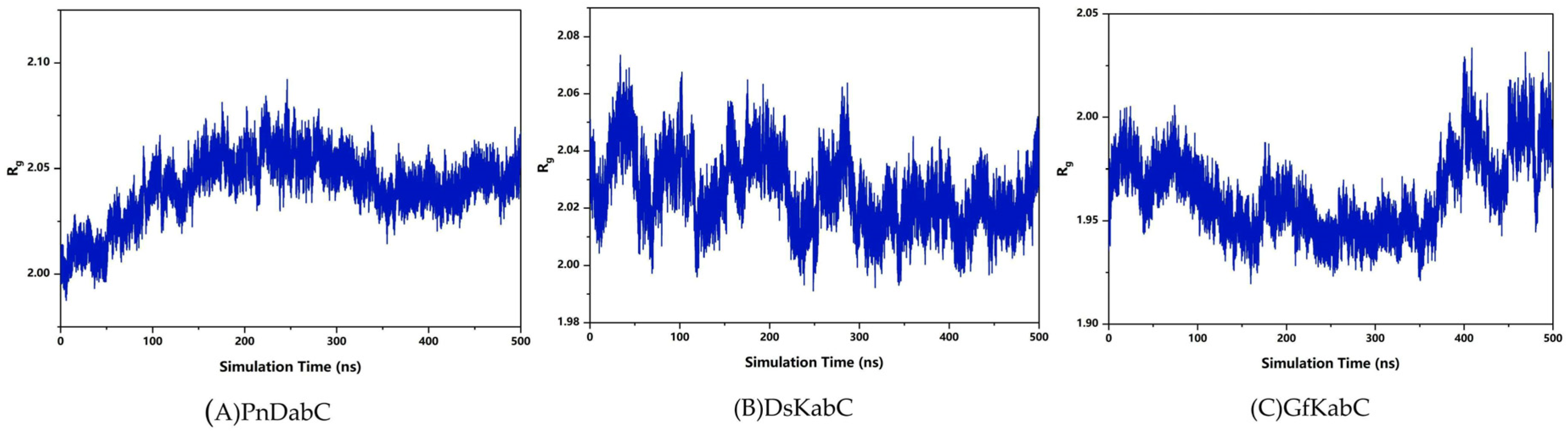
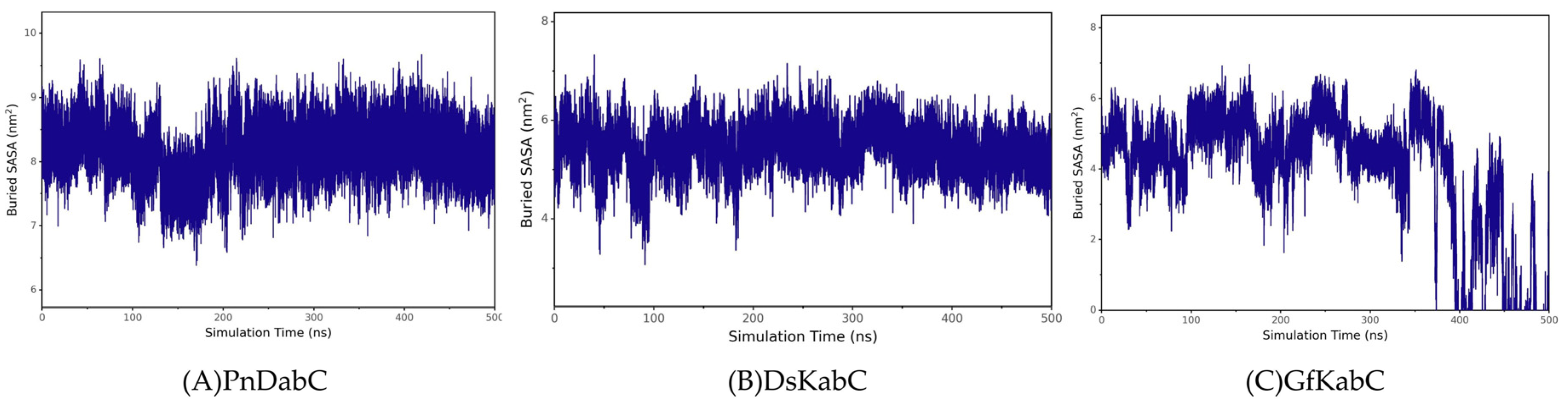
2.2. Overall Stability of the Kainoid Synthase–Substrates Complex
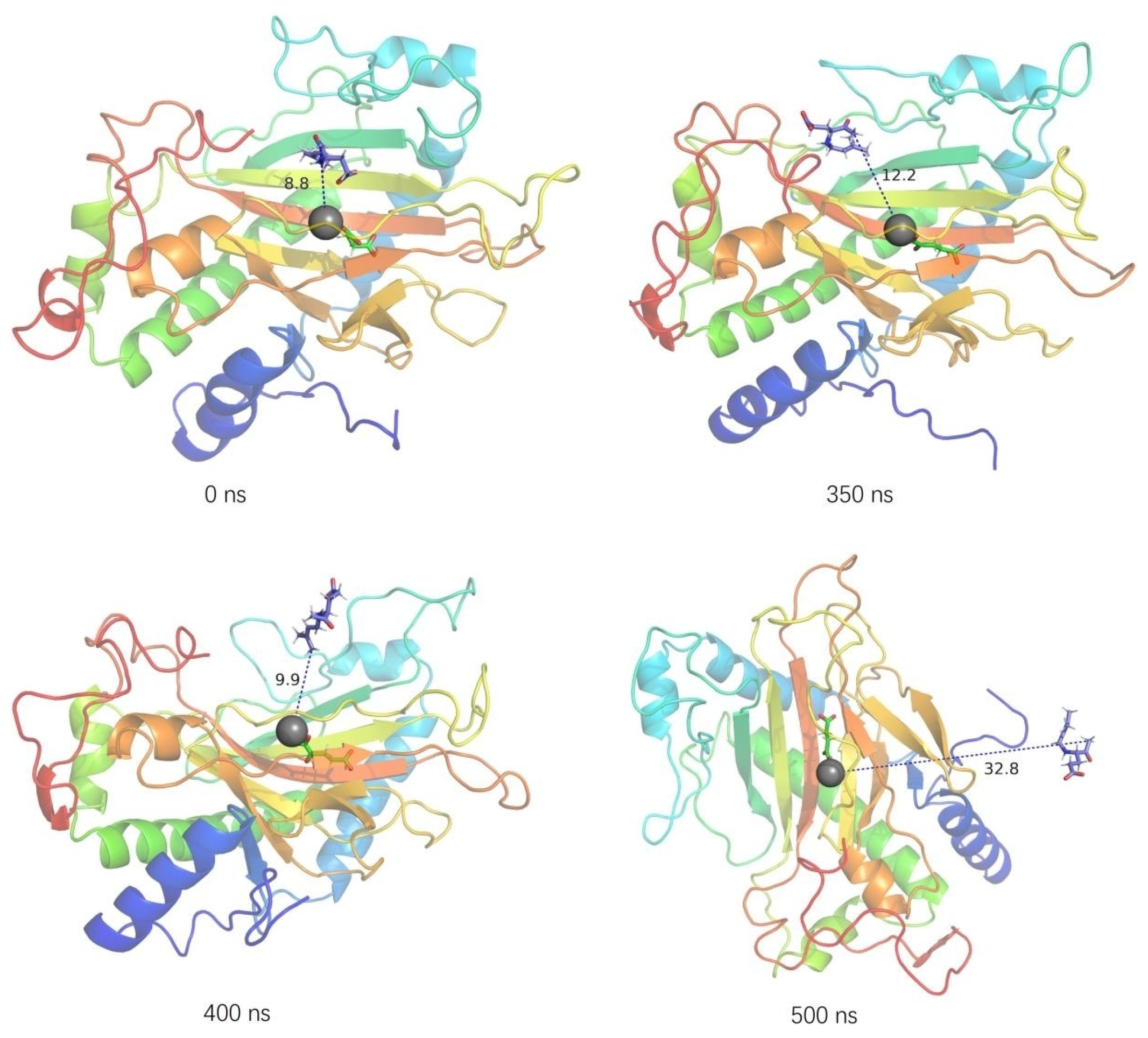
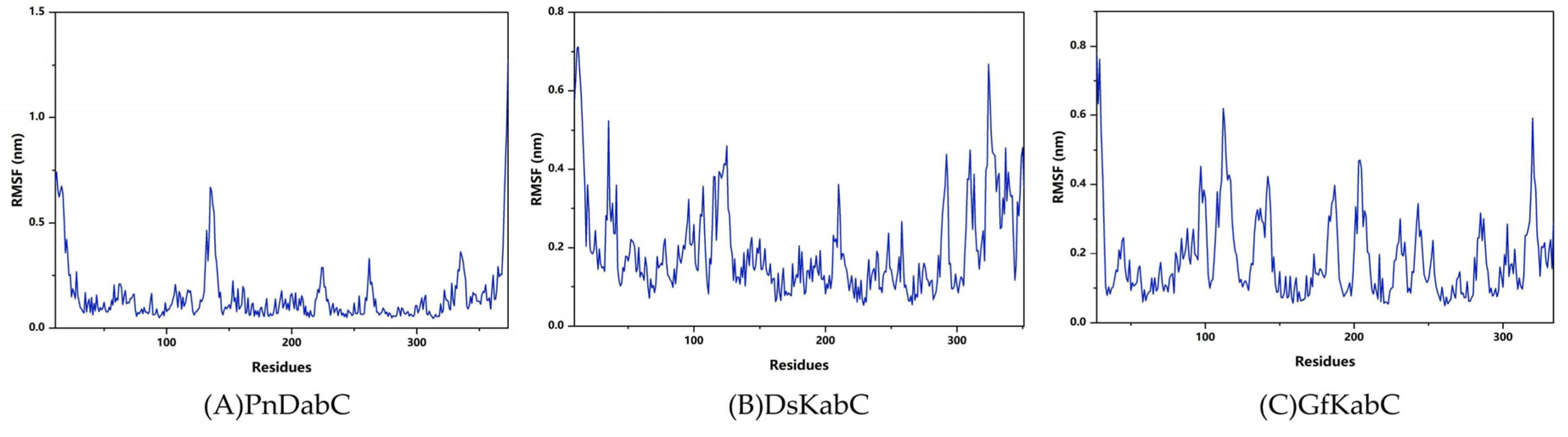
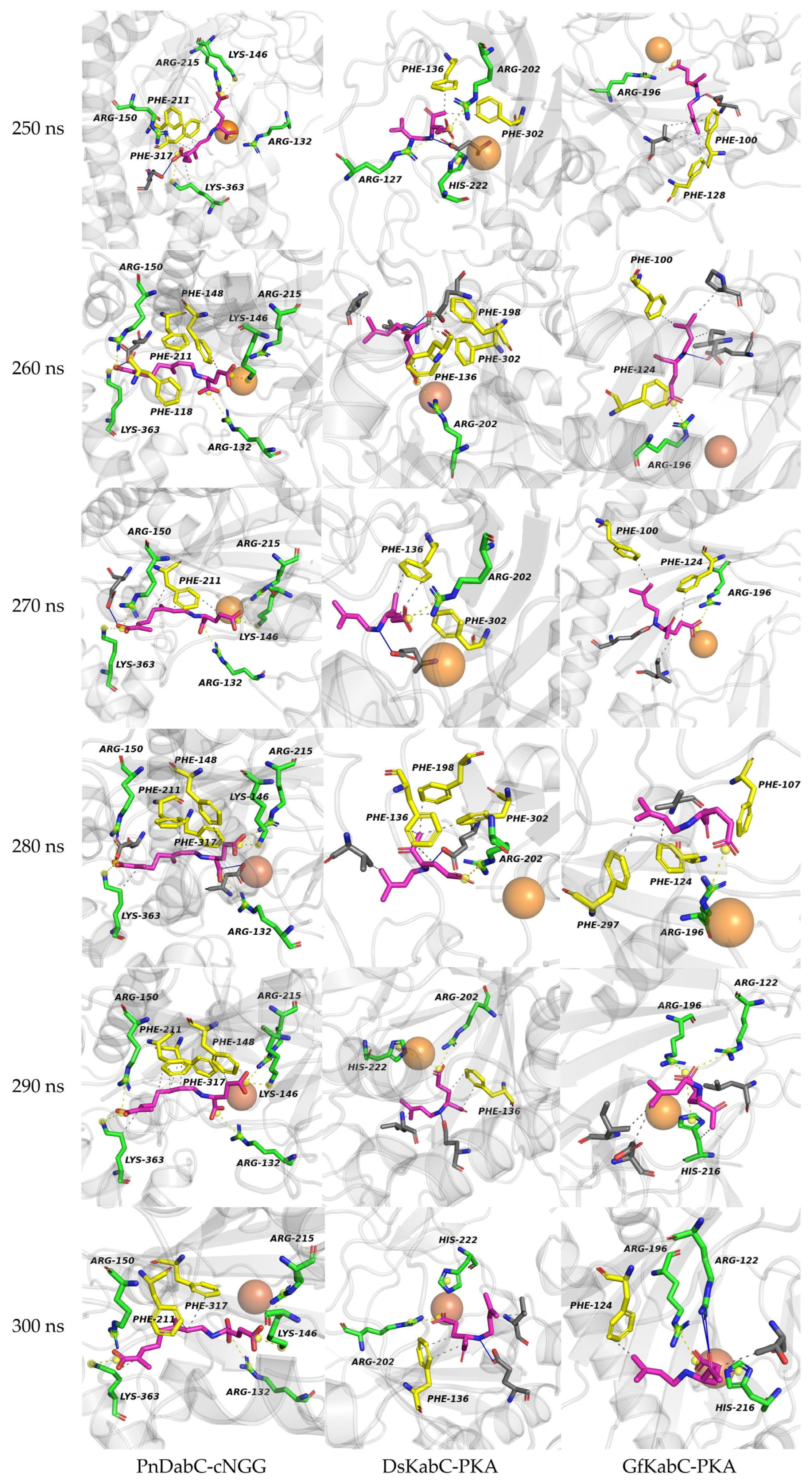
2.3. Structural Flexibility of Kainoid Synthases
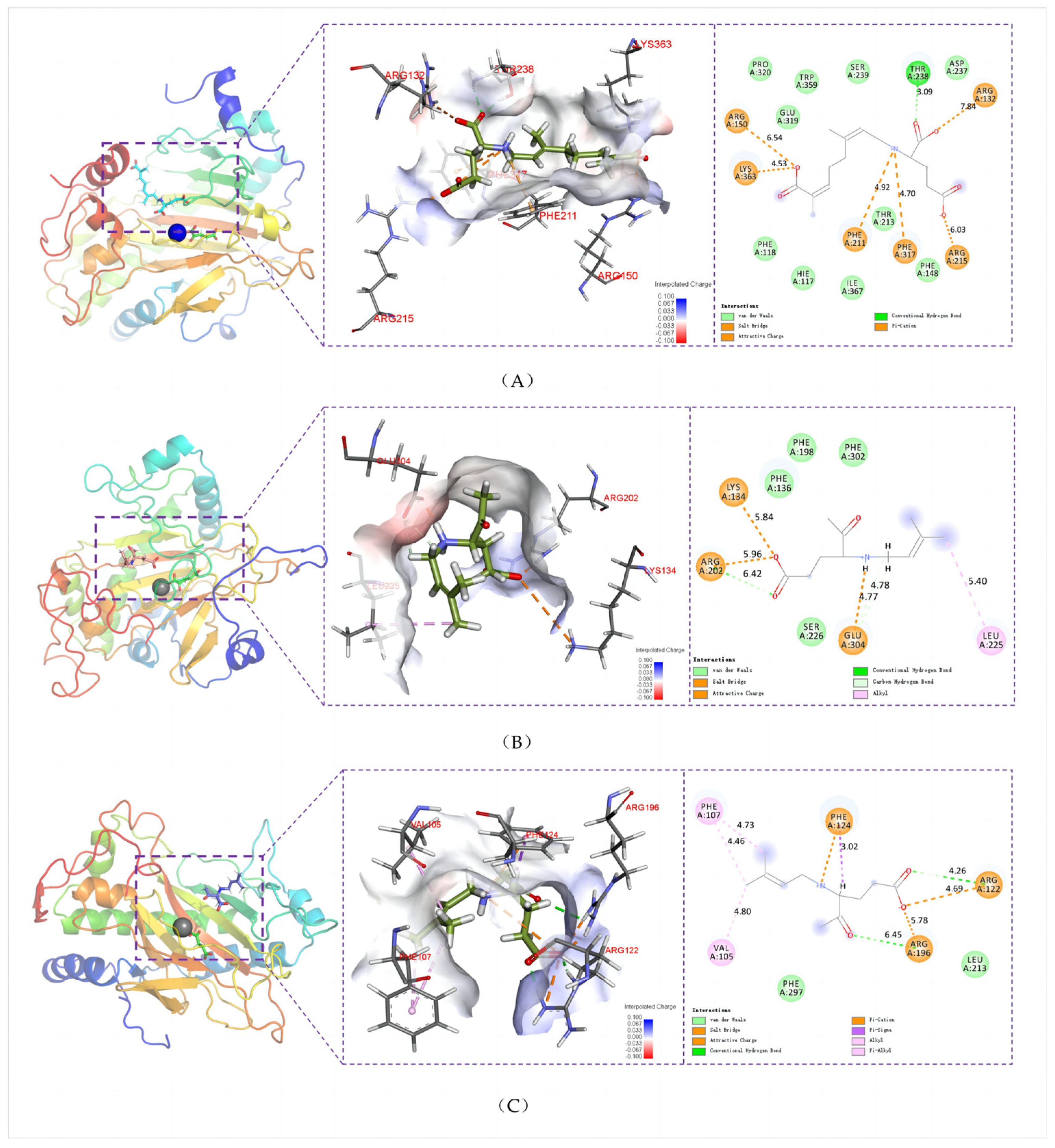
2.4. Key Structural Moieties of the Substrate Recognized by Kainoid Synthases
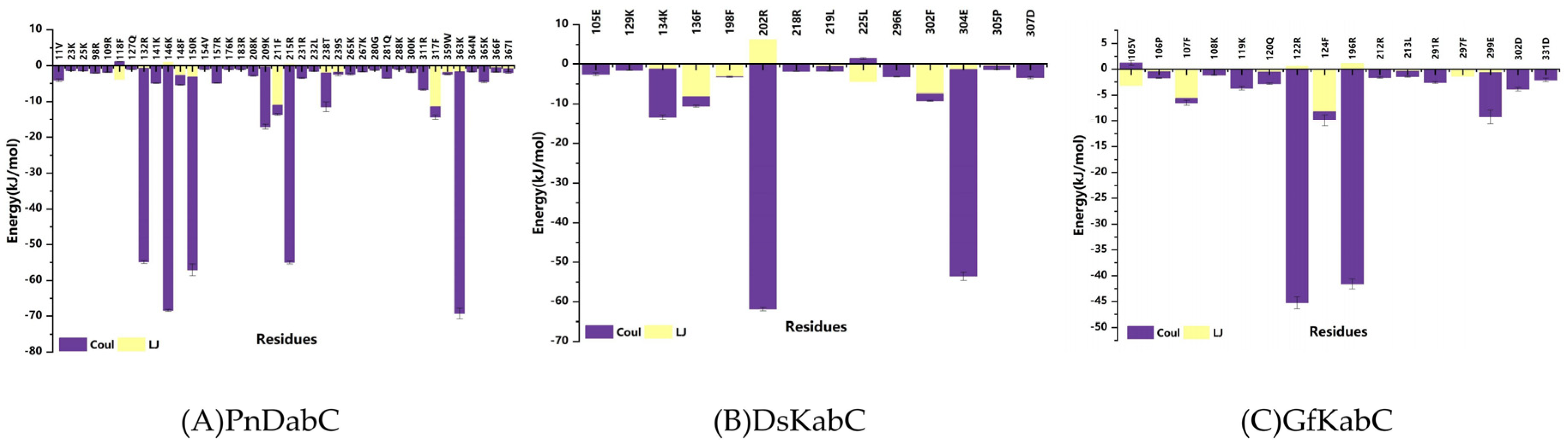
2.5. Key Residues Responsible for Interacting with the Substrates
2.6. Binding Energy Analysis of Kainoid Synthases
3. Materials and Methods
3.1. Protein Preparation and Evaluation
3.2. Molecular Dynamics Simulations
3.3. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Nie, Z.-Y.; Long, X.-P.; Bouroubi, N.E.; Liu, H.-C.; Cao, S.-T.; Chen, Y.-X.; Zheng, X.-F.; Xia, J.-L. Biosynthesis and Detection of Domoic Acid from Diatom Pseudo-nitzschia: A Review. Curr. Pharm. Biotechnol. 2023, 24, 599–610. [Google Scholar] [PubMed]
- Murakami, S.; Takemoto, T.; Shimizu, Z. Studies on the effective principles of Digenea-simplex aq. 1. separation of the effective fraction by liquid chromatography. Yakugaku Zasshi-J. Pharm. Soc. Jpn. 1953, 73, 1026–1028. [Google Scholar] [CrossRef]
- Zabaglo, K.; Chrapusta, E.; Bober, B.; Kaminski, A.; Adamski, M.; Bialczyk, J. Environmental roles and biological activity of domoic acid: A review. Algal Res. 2016, 13, 94–101. [Google Scholar] [CrossRef]
- Sperk, G. Kainic acid seizures in the rat. Prog. Neurobiol. 1994, 42, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Schwarcz, R. Lesion of striatal neurons with kainic acid provides a model for Huntington’s chorea. Nature 1976, 263, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Steele, T.S.; Brunson, J.K.; Maeno, Y.; Terada, R.; Allen, A.E.; Yotsu-Yamashita, M.; Chekan, J.R.; Moore, B.S. Domoic acid biosynthesis in the red alga Chondria armata suggests a complex evolutionary history for toxin production. Proc. Natl. Acad. Sci. USA 2022, 119, e2117407119. [Google Scholar] [CrossRef] [PubMed]
- Brunson, J.K.; McKinnie, S.M.; Chekan, J.R.; McCrow, J.P.; Miles, Z.D.; Bertrand, E.M.; Bielinski, V.A.; Luhavaya, H.; Oborník, M.; Smith, G.J. Biosynthesis of the neurotoxin domoic acid in a bloom-forming diatom. Science 2018, 361, 1356–1358. [Google Scholar] [CrossRef]
- Chekan, J.R.; McKinnie, S.M.; Moore, M.L.; Poplawski, S.G.; Michael, T.P.; Moore, B.S. Scalable biosynthesis of the seaweed neurochemical, kainic acid. Angew. Chem. Int. Ed. 2019, 58, 8454–8457. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Miyano, N.; Yashiro, S.; Umezawa, T.; Matsuda, F. Total synthesis of (−)-kainic acid and (+)-allo-kainic acid through SmI 2-mediated intramolecular coupling between allyl chloride and an α, β-unsaturated ester. Org. Biomol. Chem. 2017, 15, 6557–6566. [Google Scholar] [CrossRef]
- Nishizawa, S.; Ouchi, H.; Suzuki, H.; Ohnishi, T.; Sasaki, S.; Oyagi, Y.; Kanakogi, M.; Matsumura, Y.; Nakagawa, S.; Asakawa, T. Total synthesis of (−)-domoic acid, a potent ionotropic glutamate receptor agonist and the key compound in oceanic harmful algal blooms. Org. Biomol. Chem. 2023, 21, 1653–1656. [Google Scholar] [CrossRef]
- Chen, N.; Zhang, F.; Wu, R.; Hess Jr, B.A. Biosynthesis of Spinosyn A: A [4 + 2] or [6 + 4] Cycloaddition? ACS Catal. 2018, 8, 2353–2358. [Google Scholar] [CrossRef]
- Tantillo, D.J. Biosynthesis via carbocations: Theoretical studies on terpene formation. Nat. Prod. Rep. 2011, 28, 1035–1053. [Google Scholar] [CrossRef] [PubMed]
- Freud, Y.; Ansbacher, T.; Major, D.T. Catalytic control in the facile proton transfer in taxadiene synthase. ACS Catal. 2017, 7, 7653–7657. [Google Scholar] [CrossRef]
- Tantillo, D.J. Walking in the woods with quantum chemistry–applications of quantum chemical calculations in natural products research. Nat. Prod. Rep. 2013, 30, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, R.P.; Ho, K.C.; Tantillo, D.J. Modulation of inherent dynamical tendencies of the bisabolyl cation via preorganization in epi-isozizaene synthase. Chem. Sci. 2015, 6, 2347–2353. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.R.; Pemberton, R.P.; Tantillo, D.J. Navigating past a fork in the road: Carbocation–π interactions can manipulate dynamic behavior of reactions facing post-transition-state bifurcations. J. Am. Chem. Soc. 2017, 139, 7485–7493. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.C.; Zi, J.; Hong, Y.J.; Schulte, S.; Malchow, B.; Tantillo, D.J.; Peters, R.J. Blocking deprotonation with retention of aromaticity in a plant ent-copalyl diphosphate synthase leads to product rearrangement. Angew. Chem. Int. Ed. 2016, 55, 634–638. [Google Scholar] [CrossRef]
- Hess, B.A.; Smentek, L. Concerted, highly asynchronous, enzyme-catalyzed [4 + 2] cycloaddition in the biosynthesis of spinosyn A; computational evidence. Org. Biomol. Chem. 2012, 10, 7503–7509. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.-X.; Eriksson, L.A. Catalytic mechanism and product specificity of oxidosqualene-lanosterol cyclase: A QM/MM study. J. Phys. Chem. B 2012, 116, 13857–13862. [Google Scholar] [CrossRef]
- Zhang, F.; An, T.; Tang, X.; Zi, J.; Luo, H.-B.; Wu, R. Enzyme promiscuity versus fidelity in two sesquiterpene cyclases (TEAS versus ATAS). ACS Catal. 2019, 10, 1470–1484. [Google Scholar] [CrossRef]
- Chen, T.-Y.; Xue, S.; Tsai, W.-C.; Chien, T.-C.; Guo, Y.; Chang, W. Deciphering pyrrolidine and olefin formation mechanism in kainic acid biosynthesis. ACS Catal. 2021, 11, 278–282. [Google Scholar] [CrossRef]
- Hopiavuori, A.R.; Huffman, R.T.; McKinnie, S.M.K. Expression, purification, and biochemical characterization of micro- and macroalgal kainoid synthases. Methods Enzymol. 2024, in press. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Wu, X.; Xiu, Z.; Wang, J.; Yin, L.; Li, G. Understanding the molecular mechanism of binding modes of Aurora A inhibitors by long time scale GPU dynamics. J. Theor. Comput. Chem. 2013, 12, 1341003. [Google Scholar] [CrossRef]
- Xia, Y.; Guo, W.; Han, L.; Shen, W.; Chen, X.; Yang, H. Significant improvement of both catalytic efficiency and stability of fructosyltransferase from Aspergillus niger by structure-guided engineering of key residues in the conserved sequence of the catalytic domain. J. Agric. Food Chem. 2022, 70, 7202–7210. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Kardam, V.; Tripathi, A.; TG, S.; Dubey, K.D. The performance of different water models on the structure and function of cytochrome P450 enzymes. J. Chem. Inf. Model. 2022, 62, 6679–6690. [Google Scholar] [CrossRef] [PubMed]
- Ataei, Z.; Nouri, Z.; Tavakoli, F.; Pourreza, M.R.; Narrei, S.; Tabatabaiefar, M.A. Novel in-frame duplication variant characterization in late infantile metachromatic leukodystrophy using whole-exome sequencing and molecular dynamics simulation. PLoS ONE 2023, 18, e0282304. [Google Scholar] [CrossRef]
- Li, Z.-L.; Buck, M. Modified potential functions result in enhanced predictions of a protein complex by all-atom molecular dynamics simulations, confirming a stepwise association process for native protein–protein interactions. J. Chem. Theory Comput. 2019, 15, 4318–4331. [Google Scholar] [CrossRef]
- Li, S.; Cao, L.; Yang, X.; Wu, X.; Xu, S.; Liu, Y. Simultaneously optimizing multiple properties of β-glucosidase Bgl6 using combined (semi-) rational design strategies and investigation of the underlying mechanisms. Bioresour. Technol. 2023, 374, 128792. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Area (Å2) | Vol. (Å3) | Avg Dep.(Å) | Max Dep.(Å) | Avg Hyd. |
|---|---|---|---|---|---|
| PnDabC-cNGG | 680.81 | 376.92 | 1.67 | 4.41 | −0.02 |
| DsKabC-PKA | 667.45 | 408.46 | 1.61 | 5.5 | 0.17 |
| GfKabC-PKA | 730.44 | 438.26 | 1.47 | 4.41 | 0.15 |
| Complex | PnDabC-cNGG | DsKabC-PKA | GfKabC-PKA |
|---|---|---|---|
| ΔEvdw | −133.164 ± 6.729 | −71.61 ± 1.427 | −60.76 ± 5.734 |
| ΔEele | −411.14 ± 25.248 | −230.503 ± 3.59 | −186.169 ± 19.89 |
| ΔEpol | 641.384 ± 24.122 | 351.107 ± 4.775 | 244.753 ± 21.658 |
| ΔEnonpol | −22.355 ± 0.216 | −14.529 ± 0.031 | −13.796 ± 0.807 |
| ΔEMMPBSA | 74.724 ± 6.145 | 34.465 ± 2.689 | −15.972 ± 4.005 |
| −TΔS | 82.94 ± 6.966 | 32.564 ± 2.885 | 53.314 ± 6.549 |
| ΔGbind * | 157.664 ± 4.835 | 67.029 ± 4.931 | 37.342 ± 2.572 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Z.; Li, X.; Jiang, R.; Li, J.; Cao, F.; Sun, M.; Wang, L. Molecular Dynamics Simulation Reveal the Structure–Activity Relationships of Kainoid Synthases. Mar. Drugs 2024, 22, 326. https://doi.org/10.3390/md22070326
Fan Z, Li X, Jiang R, Li J, Cao F, Sun M, Wang L. Molecular Dynamics Simulation Reveal the Structure–Activity Relationships of Kainoid Synthases. Marine Drugs. 2024; 22(7):326. https://doi.org/10.3390/md22070326
Chicago/Turabian StyleFan, Zeyu, Xinhao Li, Ruoyu Jiang, Jinqian Li, Fangyu Cao, Mingjuan Sun, and Lianghua Wang. 2024. "Molecular Dynamics Simulation Reveal the Structure–Activity Relationships of Kainoid Synthases" Marine Drugs 22, no. 7: 326. https://doi.org/10.3390/md22070326
APA StyleFan, Z., Li, X., Jiang, R., Li, J., Cao, F., Sun, M., & Wang, L. (2024). Molecular Dynamics Simulation Reveal the Structure–Activity Relationships of Kainoid Synthases. Marine Drugs, 22(7), 326. https://doi.org/10.3390/md22070326