
Aspergillusidone G Exerts Anti-Neuroinflammatory Effects via Inhibiting MMP9 Through Integrated Bioinformatics and Experimental Analysis: Implications for Parkinson’s Disease Intervention


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
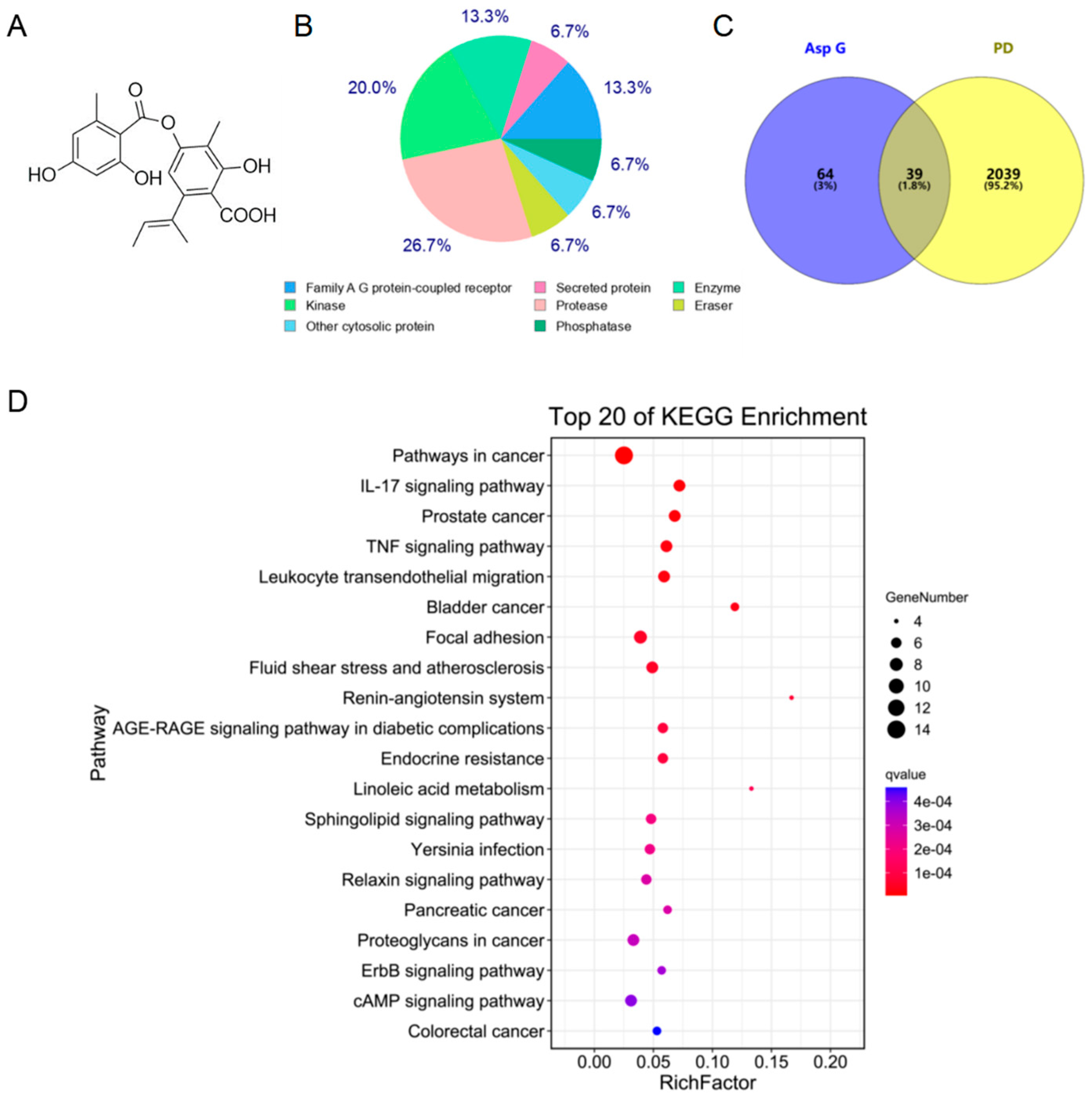
2.1. Asp G Possesses a Great Therapeutic Potential in the Inflammatory Mechanism of PD
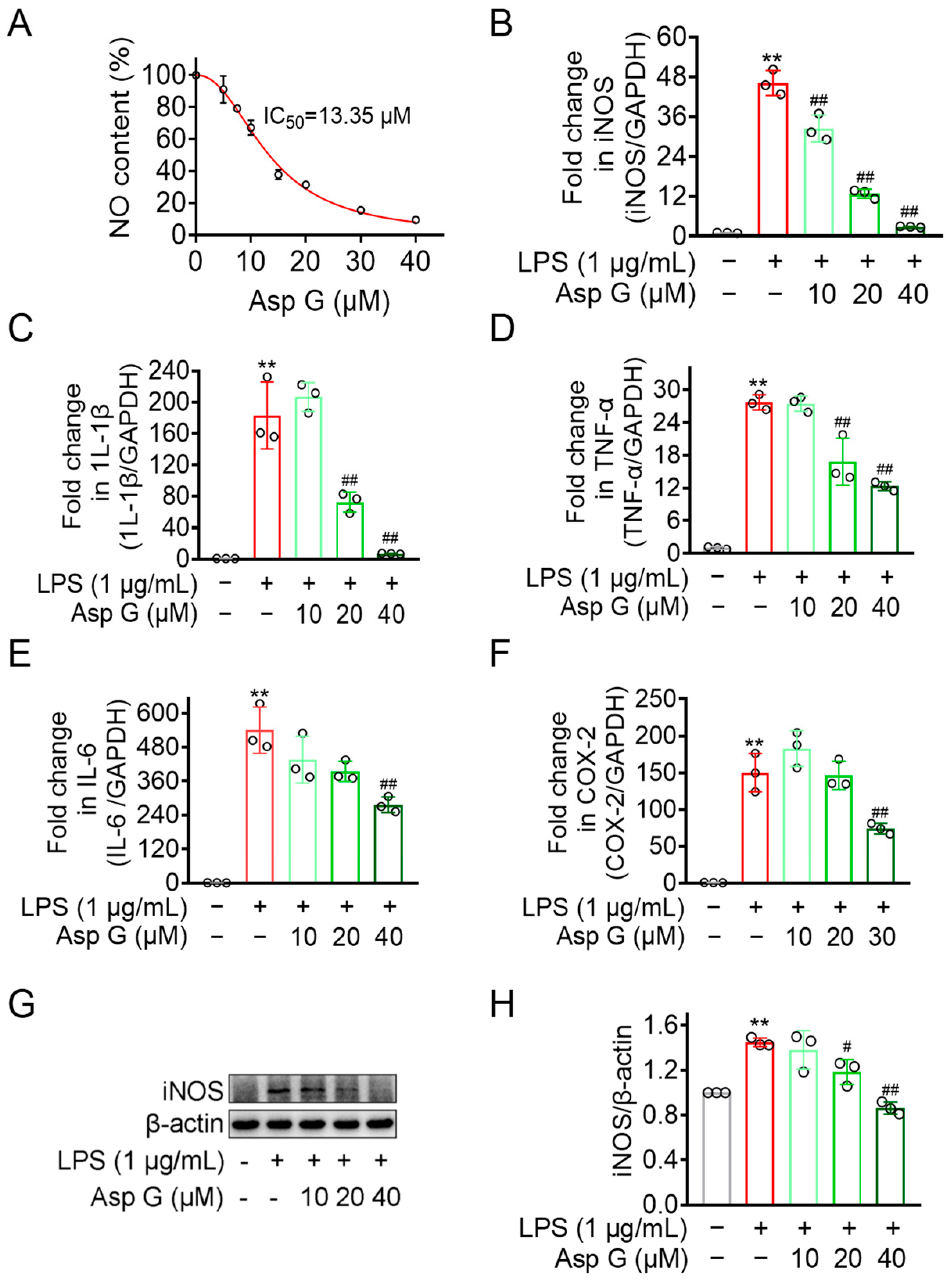
2.2. Asp G Dose-Dependently Diminishes Pro-Inflammatory Factors Expression in LPS-Induced BV2 Cells
2.3. MMP9 Was the Potential Anti-PD Target of Asp G
2.4. Asp G Directly Affected the Enzymatic Activity of the Active Form of MMP9 Induced by APMA
2.5. Asp G Ameliorated the Production of MMP9 by Regulating Its Gene and Protein Expression in LPS-Induced BV2 Microglia
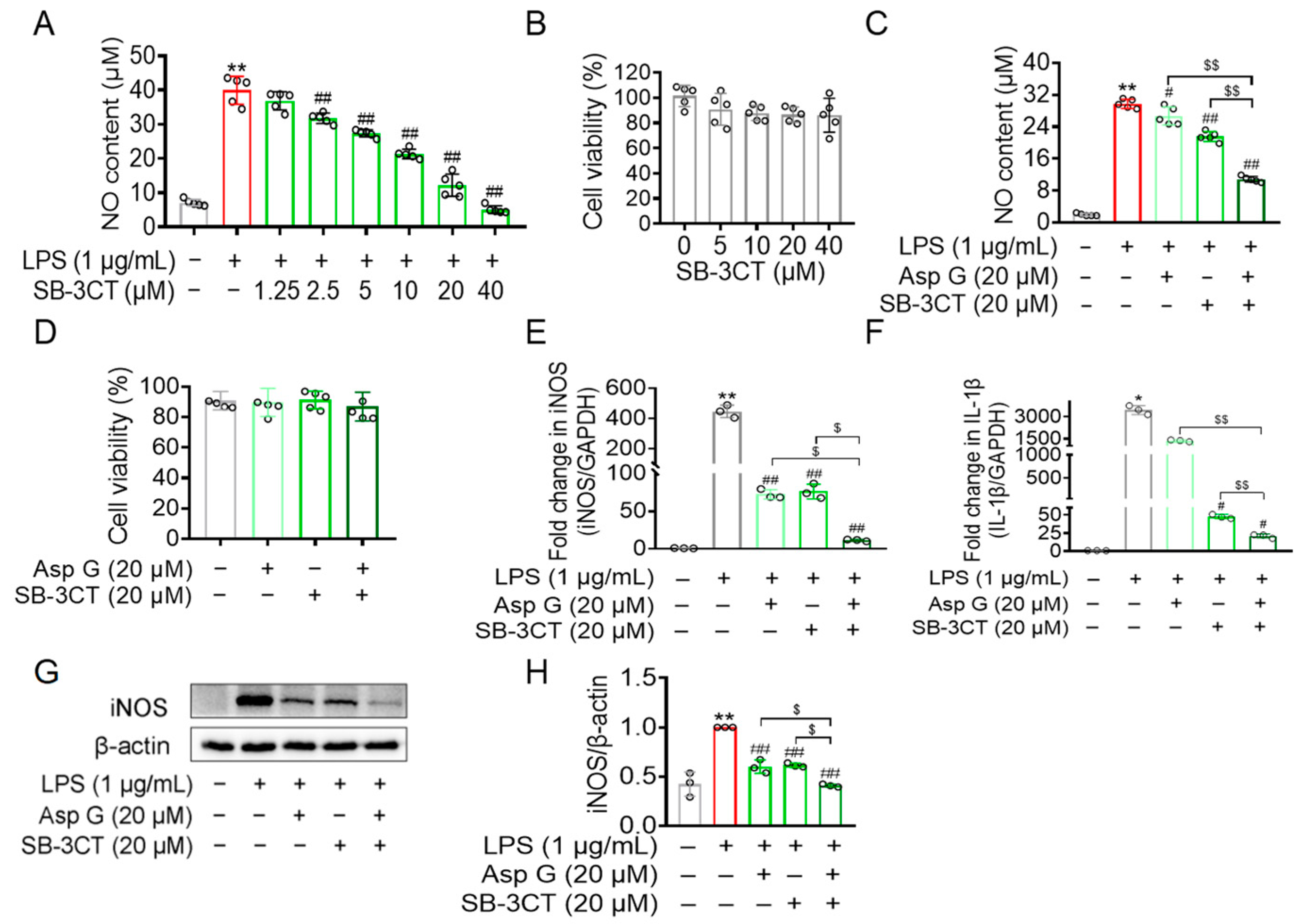
2.6. Asp G Attenuated the Production of Inflammation-Related Factors by Regulating MMP9 in LPS-Induced BV2 Microglia
3. Discussion
3.1. Anti-Neuroinflammatory Mechanism of Asp G: An Implication for PD Intervention
3.2. MMP9 Is a Potential Therapeutic Target of Asp G
4. Materials and Methods
4.1. Reagents
4.2. Cell Line and Culture Conditions
4.3. Network Pharmacology Prediction
4.4. Molecular Docking
4.5. Griess Assay
4.6. CCK-8 Assay
4.7. Quantitative Real-Time PCR (qRT-PCR)
4.8. Western Blot
4.9. Gelatin Zymography
4.10. In Vitro Activity Assay of MMP9
4.11. Statistics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Tarkang, P.A.; Appiah-Opong, R.; Ofori, M.F.; Ayong, L.S.; Nyarko, A.K. Application of multi-target phytotherapeutic concept in malaria drug discovery: A systems biology approach in biomarker identification. Biomark. Res. 2016, 4, 25. [Google Scholar] [CrossRef]
- Yarla, N.S.; Bishayee, A.; Sethi, G.; Reddanna, P.; Kalle, A.M.; Dhananjaya, B.L.; Dowluru, K.S.; Chintala, R.; Duddukuri, G.R. Targeting arachidonic acid pathway by natural products for cancer prevention and therapy. Semin. Cancer Biol. 2016, 40–41, 48–81. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.E.; Fernández, E.; Quilhot, W.; Lissi, E. Antioxidant activity of depsides and depsidones. Phytochemistry 1994, 37, 1585–1587. [Google Scholar] [CrossRef] [PubMed]
- Perlatti, B.; Lan, N.; Earp, C.E.; AghaAmiri, S.; Vargas, S.H.; Azhdarinia, A.; Bills, G.F.; Gloer, J.B. Arenicolins: C-glycosylated depsides from Penicillium arenicola. J. Nat. Prod. 2020, 83, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Sirwi, A.; Eid, B.G.; Mohamed, S.G.A.; Mohamed, G.A. Fungal depsides-naturally inspiring molecules: Biosynthesis, structural characterization, and biological activities. Metabolites 2021, 11, 683. [Google Scholar] [CrossRef]
- Ureña-Vacas, I.; González-Burgos, E.; Divakar, P.K.; Gómez-Serranillos, M.P. Lichen depsides and tridepsides: Progress in pharmacological approaches. J. Fungi 2023, 9, 116. [Google Scholar] [CrossRef]
- Lokvam, J.; Clausen, T.P.; Grapov, D.; Coley, P.D.; Kursar, T.A. Galloyl depsides of tyrosine from young leaves of Inga laurina. J. Nat. Prod. 2007, 70, 134–136. [Google Scholar] [CrossRef]
- Yang, W.C.; Bao, H.Y.; Liu, Y.Y.; Nie, Y.Y.; Yang, J.M.; Hong, P.Z.; Zhang, Y. Depsidone derivatives and a cyclopeptide produced by marine fungus Aspergillus unguis under chemical induction and by its plasma induced mutant. Molecules 2018, 23, 2245. [Google Scholar] [CrossRef]
- Morshed, M.T.; Vuong, D.; Crombie, A.; Lacey, A.E.; Karuso, P.; Lacey, E.; Piggott, A.M. Expanding antibiotic chemical space around the nidulin pharmacophore. Org. Biomol. Chem. 2018, 16, 3038–3051. [Google Scholar] [CrossRef]
- Phainuphong, P.; Rukachaisirikul, V.; Phongpaichit, S.; Sakayaroj, J.; Kanjanasirirat, P.; Borwornpinyo, S.; Akrimajirachoote, N.; Yimnual, C.; Muanprasat, C. Depsides and depsidones from the soil-derived fungus Aspergillus unguis PSU-RSPG204. Tetrahedron 2018, 74, 5691–5699. [Google Scholar] [CrossRef]
- Ban, F.; Zhou, L.; Yang, Z.; Liu, Y.; Zhang, Y. Aspergillusidone G potentiates the anti-inflammatory effects of Polaprezinc in LPS-induced BV2 microglia: A bioinformatics and experimental study. Mar. Drugs 2024, 22, 324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, W.C.; Nie, Y.Y.; Yang, Z.Y.; Liu, Y.Y.; Song, C.; Hong, P.Z. Application of the Compound Aspergillusidone G in the Preparation of Neuroprotective Drugs. CN201910808567.4, 17 March 2023. [Google Scholar]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Pal, G. Initial management of Parkinson’s disease. Bmj 2014, 349, g6258. [Google Scholar] [CrossRef]
- Moehle, M.S.; West, A.B. M1 and M2 immune activation in Parkinson’s disease: Foe and ally? Neuroscience 2015, 302, 59–73. [Google Scholar] [CrossRef]
- Gundersen, V. Parkinson’s disease: Can targeting inflammation be an effective neuroprotective strategy? Front. Neurosci. 2020, 14, 580311. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Qin, X.Y.; Zhang, S.P.; Cao, C.; Loh, Y.P.; Cheng, Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: A systematic review and meta-analysis. JAMA Neurol. 2016, 73, 1316–1324. [Google Scholar] [CrossRef]
- Starhof, C.; Winge, K.; Heegaard, N.H.H.; Skogstrand, K.; Friis, S.; Hejl, A. Cerebrospinal fluid pro-inflammatory cytokines differentiate parkinsonian syndromes. J. Neuroinflamm. 2018, 15, 305. [Google Scholar] [CrossRef]
- Sun, H.Y.; Wu, J.; Wang, R.; Zhang, S.; Xu, H.; Kaznacheyeva, E.; Lu, X.J.; Ren, H.G.; Wang, G.H. Pazopanib alleviates neuroinflammation and protects dopaminergic neurons in LPS-stimulated mouse model by inhibiting MEK4-JNK-AP-1 pathway. Acta Pharmacol. Sin. 2023, 44, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.H.; Zhang, Y.N.; Zhang, J.N.; Gu, L.; Yang, H.M.; Huang, Y.Y.; Xia, N.; Zhang, H. Urate inhibits microglia activation to protect neurons in an LPS-induced model of Parkinson’s disease. J. Neuroinflamm. 2018, 15, 131. [Google Scholar] [CrossRef]
- Nam, H.Y.; Nam, J.H.; Yoon, G.; Lee, J.Y.; Nam, Y.; Kang, H.J.; Cho, H.J.; Kim, J.; Hoe, H.S. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J. Neuroinflamm. 2018, 15, 271. [Google Scholar] [CrossRef]
- Choi, J.W.; Jo, S.W.; Kim, D.E.; Paik, I.Y.; Balakrishnan, R. Aerobic exercise attenuates LPS-induced cognitive dysfunction by reducing oxidative stress, glial activation, and neuroinflammation. Redox Biol. 2024, 71, 103101. [Google Scholar] [CrossRef] [PubMed]
- Hannocks, M.J.; Zhang, X.; Gerwien, H.; Chashchina, A.; Burmeister, M.; Korpos, E.; Song, J.; Sorokin, L. The gelatinases, MMP-2 and MMP-9, as fine tuners of neuroinflammatory processes. Matrix Biol. 2019, 75–76, 102–113. [Google Scholar] [CrossRef]
- Liu, C.Z.; Guo, D.S.; Ma, J.J.; Dong, L.R.; Chang, Q.Q.; Yang, H.Q.; Liang, K.K.; Li, X.H.; Yang, D.W.; Fan, Y.Y.; et al. Correlation of matrix metalloproteinase 3 and matrix metalloproteinase 9 levels with non-motor symptoms in patients with Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 889257. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Ruan, Z.; Zhang, D.; Liu, X.; Hou, L.; Wang, Q. Rotenone impairs learning and memory in mice through microglia-mediated blood brain barrier disruption and neuronal apoptosis. Chemosphere 2022, 291, 132982. [Google Scholar] [CrossRef]
- Zaman, B.; Mostafa, I.; Hassan, T.; Ahmed, S.; Esha, N.J.I.; Chowdhury, F.A.; Bosu, T.; Chowdhury, H.N.; Mallick, A.; Islam, M.S.; et al. Tolperisone hydrochloride improves motor functions in Parkinson’s disease via MMP-9 inhibition and by downregulating p38 MAPK and ERK1/2 signaling cascade. Biomed. Pharmacother. 2024, 174, 116438. [Google Scholar] [CrossRef]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Shrestha, S.; Kim, M.J.; Eldridge, M.; Lehmann, M.L.; Frankland, M.; Liow, J.S.; Yu, Z.X.; Cortes-Salva, M.; Telu, S.; Henter, I.D.; et al. PET measurement of cyclooxygenase-2 using a novel radioligand: Upregulation in primate neuroinflammation and first-in-human study. J. Neuroinflamm. 2020, 17, 140. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; Shastri, V.P. Matrix-metalloproteinase-9 is cleaved and activated by cathepsin K. BMC Res. Notes 2015, 8, 322. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Fliszar, C.J.; Broekelmann, T.J.; Mecham, R.P.; Senior, R.M.; Welgus, H.G. Activation of the 92-kDa gelatinase by stromelysin and 4-aminophenylmercuric acetate. Differential processing and stabilization of the carboxyl-terminal domain by tissue inhibitor of metalloproteinases (TIMP). J. Biol. Chem. 1995, 270, 6351–6356. [Google Scholar] [CrossRef] [PubMed]
- Vandooren, J.; Van den Steen, P.E.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [Google Scholar] [CrossRef]
- de Almeida, L.G.N.; Thode, H.; Eslambolchi, Y.; Chopra, S.; Young, D.; Gill, S.; Devel, L.; Dufour, A. Matrix metalloproteinases: From molecular mechanisms to physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 2022, 74, 712–768. [Google Scholar] [CrossRef]
- Ikejiri, M.; Bernardo, M.M.; Bonfil, R.D.; Toth, M.; Chang, M.; Fridman, R.; Mobashery, S. Potent mechanism-based inhibitors for matrix metalloproteinases. J. Biol. Chem. 2005, 280, 33992–34002. [Google Scholar] [CrossRef] [PubMed]
- Boguszewska-Czubara, A.; Budzynska, B.; Skalicka-Wozniak, K.; Kurzepa, J. Perspectives and new aspects of metalloproteinases’ inhibitors in the therapy of CNS disorders: From chemistry to medicine. Curr. Med. Chem. 2019, 26, 3208–3224. [Google Scholar] [CrossRef]
- Lim, D.W.; Lee, J.E.; Lee, C.; Kim, Y.T. Natural products and their neuroprotective effects in degenerative brain diseases: A comprehensive review. Int. J. Mol. Sci. 2024, 25, 11223. [Google Scholar] [CrossRef]
- Sommer, A.; Marxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 2018, 23, 123–131.e126. [Google Scholar] [CrossRef]
- Lin, C.H.; Lin, H.Y.; Ho, E.P.; Ke, Y.C.; Cheng, M.F.; Shiue, C.Y.; Wu, C.H.; Liao, P.H.; Hsu, A.Y.; Chu, L.A.; et al. Mild chronic colitis triggers parkinsonism in LRRK2 mutant mice through activating TNF-α pathway. Mov. Disord. 2022, 37, 745–757. [Google Scholar] [CrossRef]
- Choi, I.; Kim, B.; Byun, J.W.; Baik, S.H.; Huh, Y.H.; Kim, J.H.; Mook-Jung, I.; Song, W.K.; Shin, J.H.; Seo, H.; et al. LRRK2 G2019S mutation attenuates microglial motility by inhibiting focal adhesion kinase. Nat. Commun. 2015, 6, 8255. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, U.; Rosh, I.; Ben Ezer, R.; Nayak, R.; Hussein, Y.; Choudhary, A.; Djamus, J.; Manole, A.; Houlden, H.; Gage, F.H.; et al. Upregulated ECM genes and increased synaptic activity in Parkinson’s human DA neurons with PINK1/PRKN mutations. NPJ Park. Dis. 2024, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef]
- Brodacki, B.; Staszewski, J.; Toczyłowska, B.; Kozłowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Liberatore, G.T.; Jackson-Lewis, V.; Vukosavic, S.; Mandir, A.S.; Vila, M.; McAuliffe, W.G.; Dawson, V.L.; Dawson, T.M.; Przedborski, S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat. Med. 1999, 5, 1403–1409. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Annese, V.; Herrero, M.T.; Di Pentima, M.; Gomez, A.; Lombardi, L.; Ros, C.M.; De Pablos, V.; Fernandez-Villalba, E.; De Stefano, M.E. Metalloproteinase-9 contributes to inflammatory glia activation and nigro-striatal pathway degeneration in both mouse and monkey models of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. Brain Struct. Funct. 2015, 220, 703–727. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, Y.; Sun, Q.; Wang, R.; Lu, H.; Zhang, X.; Gao, L.; Cai, Q.; Liu, B.; Deng, G. Self-assembled genistein nanoparticles suppress the epithelial-mesenchymal transition in glioblastoma by targeting MMP9. Mater. Today Bio 2025, 31, 101606. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Zhang, D.; Huang, R.; Sun, W.; Hou, L.; Zhao, J.; Wang, Q. Microglial activation damages dopaminergic neurons through MMP-2/-9-mediated increase of blood-brain barrier permeability in a Parkinson’s disease mouse model. Int. J. Mol. Sci. 2022, 23, 2793. [Google Scholar] [CrossRef] [PubMed]
- Si, X.; Dai, S.; Fang, Y.; Tang, J.; Wang, Z.; Li, Y.; Song, Z.; Chen, Y.; Liu, Y.; Zhao, G.; et al. Matrix metalloproteinase-9 inhibition prevents aquaporin-4 depolarization-mediated glymphatic dysfunction in Parkinson’s disease. J. Adv. Res. 2024, 56, 125–136. [Google Scholar] [CrossRef]
- O’Connell, J.P.; Willenbrock, F.; Docherty, A.J.; Eaton, D.; Murphy, G. Analysis of the role of the COOH-terminal domain in the activation, proteolytic activity, and tissue inhibitor of metalloproteinase interactions of gelatinase B. J. Biol. Chem. 1994, 269, 14967–14973. [Google Scholar] [CrossRef]
- Okada, Y.; Gonoji, Y.; Naka, K.; Tomita, K.; Nakanishi, I.; Iwata, K.; Yamashita, K.; Hayakawa, T. Matrix metalloproteinase 9 (92-kDa gelatinase/type IV collagenase) from HT 1080 human fibrosarcoma cells. Purification and activation of the precursor and enzymic properties. J. Biol. Chem. 1992, 267, 21712–21719. [Google Scholar] [CrossRef]
- Lee, E.J.; Ko, H.M.; Jeong, Y.H.; Park, E.M.; Kim, H.S. β-Lapachone suppresses neuroinflammation by modulating the expression of cytokines and matrix metalloproteinases in activated microglia. J. Neuroinflamm. 2015, 12, 133. [Google Scholar] [CrossRef]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Zu, G.; Sun, K.; Li, L.; Zu, X.; Han, T.; Huang, H. Mechanism of quercetin therapeutic targets for Alzheimer disease and type 2 diabetes mellitus. Sci. Rep. 2021, 11, 22959. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Kushairi, N.; Phan, C.W.; Sabaratnam, V.; David, P.; Naidu, M. Lion’s mane mushroom, Hericium erinaceus (Bull.: Fr.) Pers. suppresses H(2)O(2)-induced oxidative damage and LPS-induced inflammation in HT22 hippocampal neurons and BV2 microglia. Antioxidants 2019, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Zhu, Y.; He, G.; Ni, H.; Liu, C.; Ma, L.; Zhang, L.; Shi, D. Dexmedetomidine attenuates neuroinflammation In LPS-stimulated BV2 microglia cells through upregulation of miR-340. Drug Des. Devel Ther. 2019, 13, 3465–3475. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Wu, J.; Liu, R.; Wang, S.; Luo, J.; Yang, Y.; Qin, Y.; Li, T.; Zheng, X.; Song, J.; et al. A novel compound DBZ ameliorates neuroinflammation in LPS-stimulated microglia and ischemic stroke rats: Role of Akt(Ser473)/GSK3β(Ser9)-mediated Nrf2 activation. Redox Biol. 2020, 36, 101644. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Vandooren, J.; Knoops, S.; Aldinucci Buzzo, J.L.; Boon, L.; Martens, E.; Opdenakker, G.; Kolaczkowska, E. Differential inhibition of activity, activation and gene expression of MMP-9 in THP-1 cells by azithromycin and minocycline versus bortezomib: A comparative study. PLoS ONE 2017, 12, e0174853. [Google Scholar] [CrossRef]
- Ugarte-Berzal, E.; Martens, E.; Boon, L.; Vandooren, J.; Blockmans, D.; Proost, P.; Opdenakker, G. EDTA/gelatin zymography method to identify C1s versus activated MMP-9 in plasma and immune complexes of patients with systemic lupus erythematosus. J. Cell Mol. Med. 2019, 23, 576–585. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ban, F.; Zhou, L.; Yang, Z.; Liu, Y.; Zhang, Y. Aspergillusidone G Exerts Anti-Neuroinflammatory Effects via Inhibiting MMP9 Through Integrated Bioinformatics and Experimental Analysis: Implications for Parkinson’s Disease Intervention. Mar. Drugs 2025, 23, 181. https://doi.org/10.3390/md23050181
Ban F, Zhou L, Yang Z, Liu Y, Zhang Y. Aspergillusidone G Exerts Anti-Neuroinflammatory Effects via Inhibiting MMP9 Through Integrated Bioinformatics and Experimental Analysis: Implications for Parkinson’s Disease Intervention. Marine Drugs. 2025; 23(5):181. https://doi.org/10.3390/md23050181
Chicago/Turabian StyleBan, Fangfang, Longjian Zhou, Zhiyou Yang, Yayue Liu, and Yi Zhang. 2025. "Aspergillusidone G Exerts Anti-Neuroinflammatory Effects via Inhibiting MMP9 Through Integrated Bioinformatics and Experimental Analysis: Implications for Parkinson’s Disease Intervention" Marine Drugs 23, no. 5: 181. https://doi.org/10.3390/md23050181
APA StyleBan, F., Zhou, L., Yang, Z., Liu, Y., & Zhang, Y. (2025). Aspergillusidone G Exerts Anti-Neuroinflammatory Effects via Inhibiting MMP9 Through Integrated Bioinformatics and Experimental Analysis: Implications for Parkinson’s Disease Intervention. Marine Drugs, 23(5), 181. https://doi.org/10.3390/md23050181