The Chemistry and Pharmacology of Anatoxin-a and Related Homotropanes with respect to Nicotinic Acetylcholine Receptors

Abstract
:Introduction
The Nicotinic Acetylcholine Receptor (nAChR)
The Homotropane Alkaloids
Isolation, Biosynthesis and Analytical Methods
Homotropane Alkaloids as Nicotinic Ligands
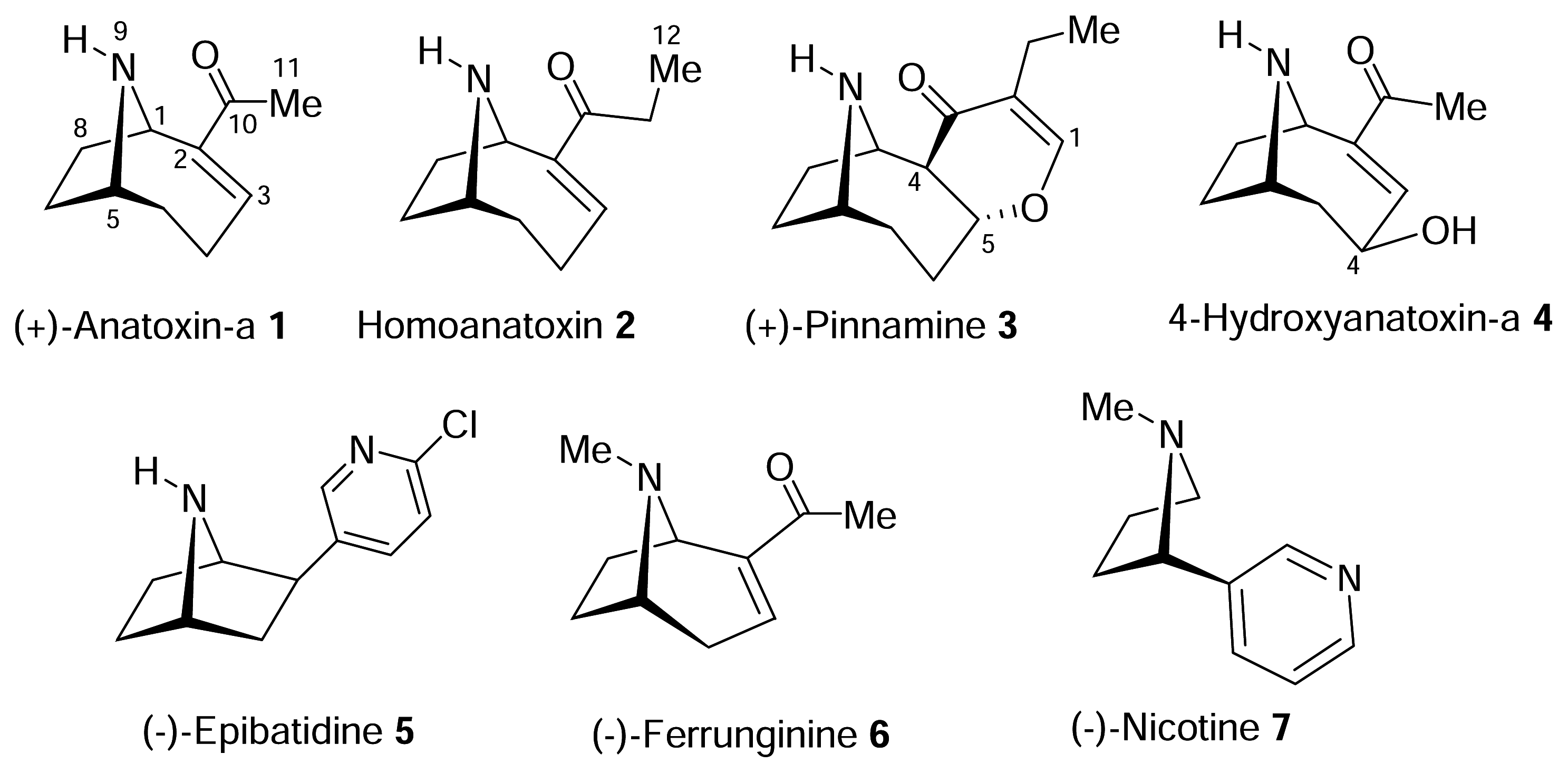
The structure of the homotropane alkaloids
Understanding the relationship between chemical structure and pharmacological profile. The nicotinic pharmacophore
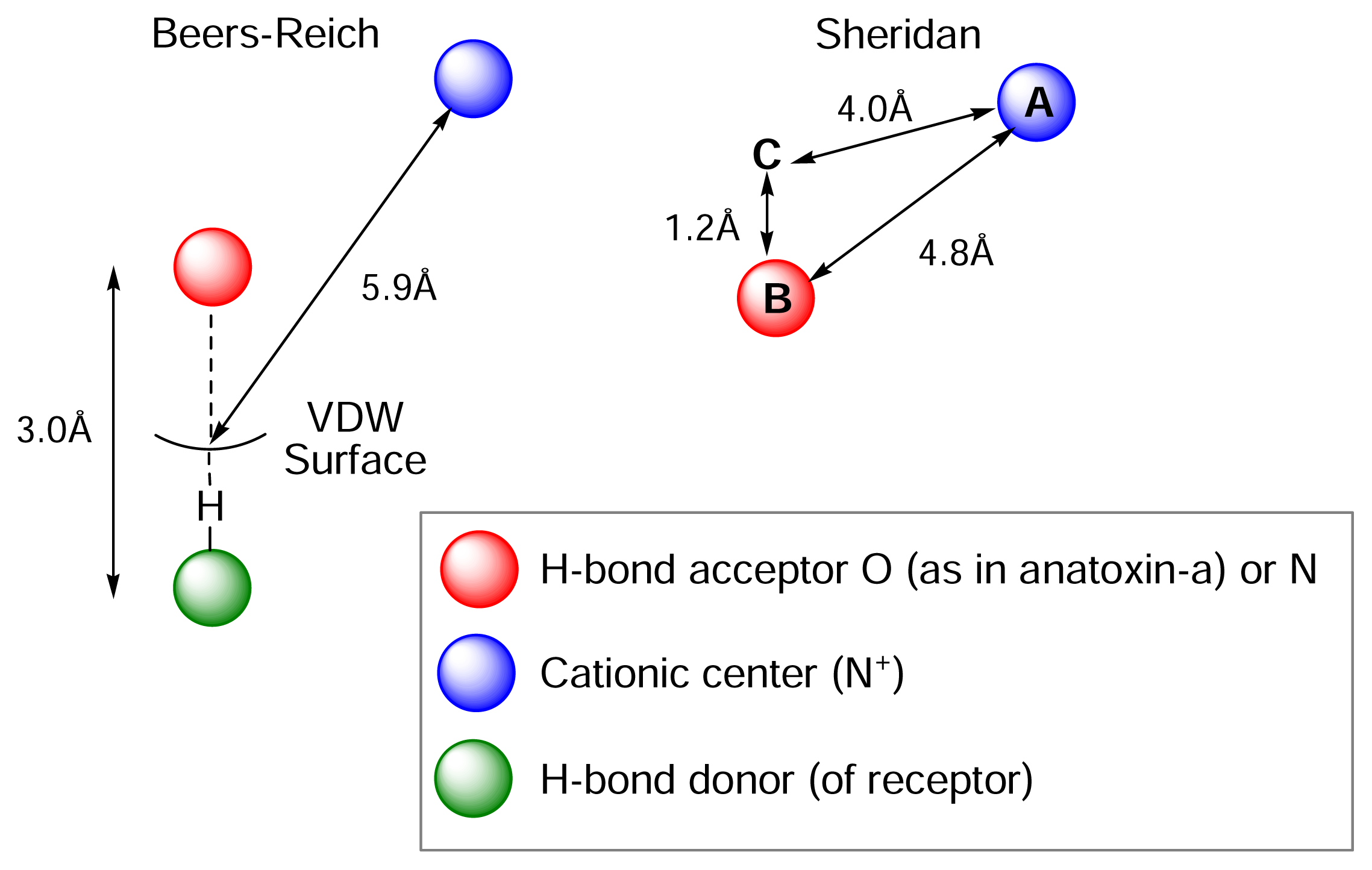
 ) (ii) an electronegative element ((
) (ii) an electronegative element ((
 ) again O or N, capable of accepting a hydrogen bond and (iii) a “dummy” site or atom (C) along which the hydrogen bond may form. In nicotine, for example, this dummy site (C) was the center of the pyridine ring, while in anatoxin-a C is the carbon of the carbonyl group. This triangular relationship again served to define the spatial relationship between the critical ligand associated sites, and Sheridan’s model required a ligand to meet key distance parameters: (
-
= 4.8 ±0.3 Ǻ; (
-C = 4.0 ±0.3 Ǻ;
-C = 1.2 Ǻ. Sheridan also suggested a “Beer-Reich distance” of 5.9 Ǻ, although given that both groups had used the same molecules and considered the same essential structural features, this level of agreement is not perhaps surprising.
) again O or N, capable of accepting a hydrogen bond and (iii) a “dummy” site or atom (C) along which the hydrogen bond may form. In nicotine, for example, this dummy site (C) was the center of the pyridine ring, while in anatoxin-a C is the carbon of the carbonyl group. This triangular relationship again served to define the spatial relationship between the critical ligand associated sites, and Sheridan’s model required a ligand to meet key distance parameters: (
-
= 4.8 ±0.3 Ǻ; (
-C = 4.0 ±0.3 Ǻ;
-C = 1.2 Ǻ. Sheridan also suggested a “Beer-Reich distance” of 5.9 Ǻ, although given that both groups had used the same molecules and considered the same essential structural features, this level of agreement is not perhaps surprising.Chemistry and Pharmacology of the Homotropane Alkaloids
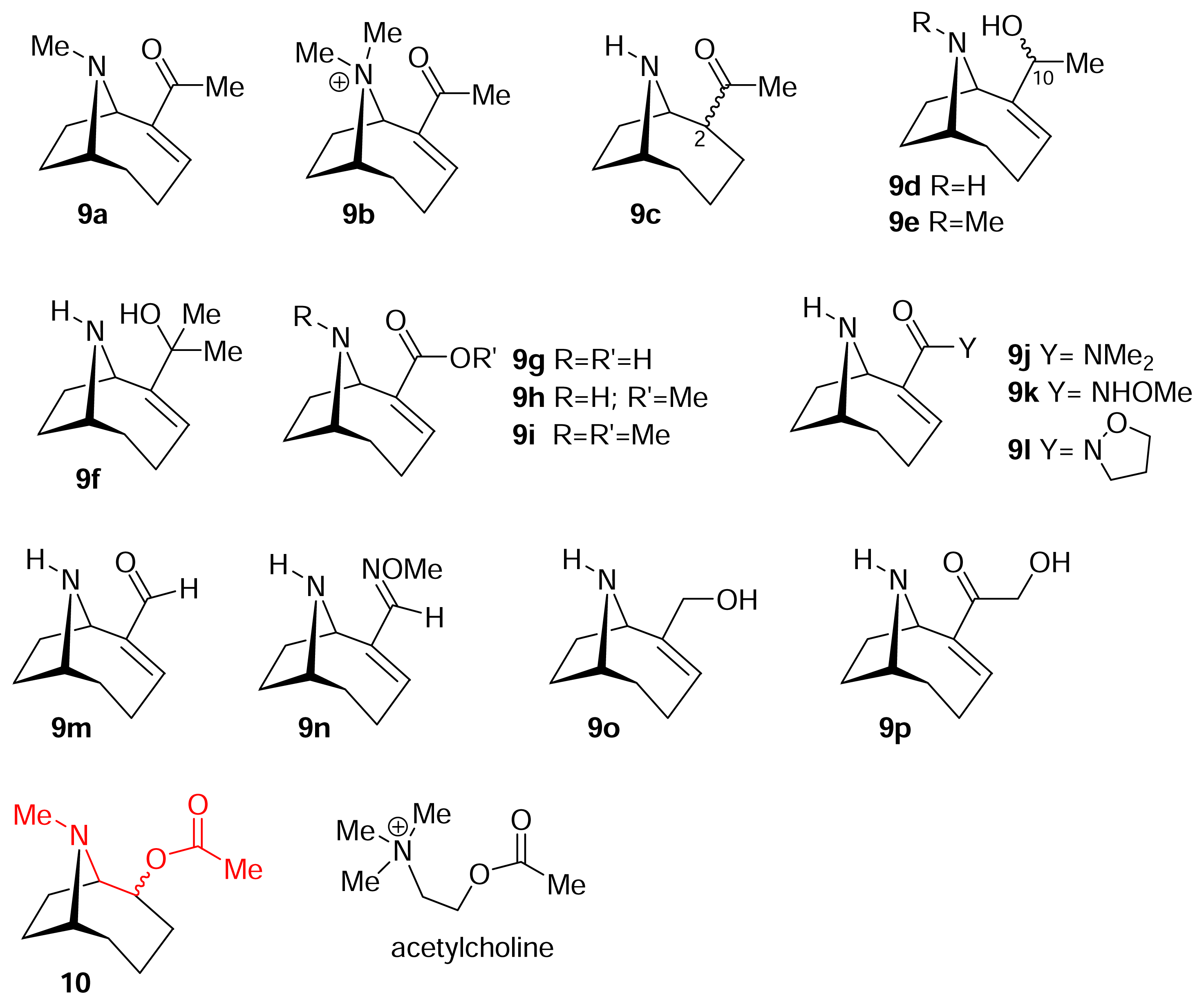
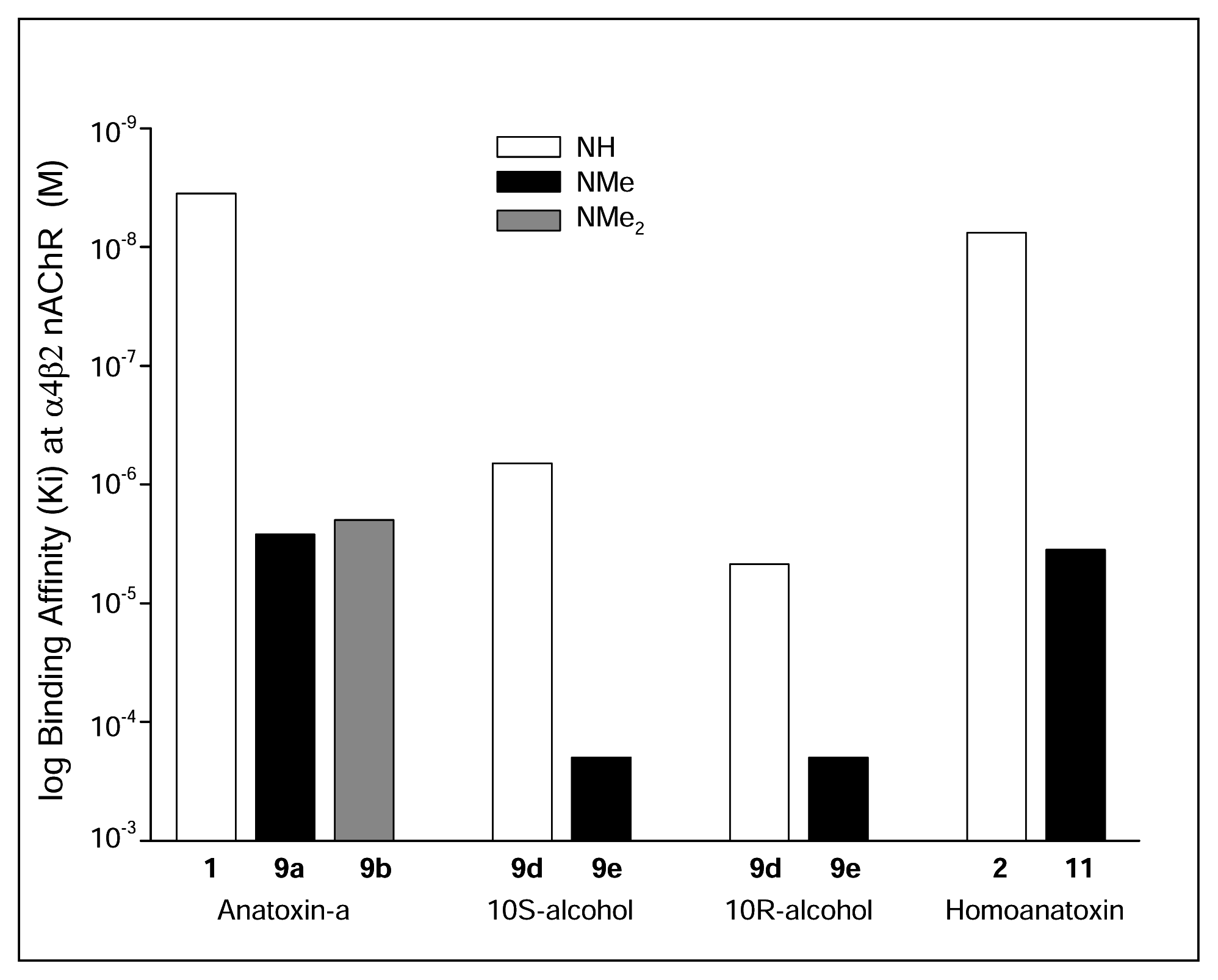
I. Structural modifications to the periphery of anatoxin-a
II. S-Cis vs. S-Trans. What is the bioactive conformation of anatoxin-a?
III. Anatoxin-a Hybrids: UB-165
Conclusions
Abbreviations
| nAChR | nicotinic acetylcholine receptors |
| PHT | pyrido[3,4-b]-homotropane |
| QSAR | quantitative structure activity relationship |
| VDW | van der Waals |
- Samples Availability: Not available.
References
- Francis, G. Poisonous Australian Lake. Nature 1878, 11–12. [Google Scholar]
- James, K. J.; Crowley, J.; Hamilton, B.; Lehane, M.; Skulberg, O.; Furey, A. Anatoxins and degradation products, determined using hybrid quadrupole time-of-flight and quadrupole ion-trap mass spectrometry: forensic investigations of cyanobacterial neurotoxin poisoning. Rapid Commun. Mass Spectrom 2005, 19, 1167–1175. [Google Scholar]
- James, K. J.; Furey, A.; Sherlock, I. R.; Stack, M. A.; Twohig, M.; Caudwell, F. B.; Skulberg, O. M. Sensitive determination of anatoxin-a, homoanatoxin-a and their degradation products by liquid chromatography with fluorimetric detection. J. Chromatogr. A 1998, 798, 147–157. [Google Scholar]
- Drager, B. Analysis of tropane and related alkaloids. J. Chromatogr. A 2002, 978, 1–35. [Google Scholar]
- Pravda, M.; Kreuzer, M. P.; Guilbault, G. G. Analysis of important freshwater and marine toxins. Anal. Lett 2002, 35, 1–15. [Google Scholar]
- Mansell, H. L. Synthetic approaches to anatoxin-a. Tetrahedron 1996, 52, 6025–6061. [Google Scholar]
- Karlin, A. Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci 2002, 3, 102–114. [Google Scholar]
- Spivak, C. E.; Witkop, B.; Albuquerque, E. X. Anatoxin-a: a novel, potent agonist at the nicotinic receptor. Mol. Pharmacol 1980, 18, 384–394. [Google Scholar]
- Carmichael, W. W.; Biggs, D. F.; Gorham, P. R. Toxicology and Pharmacological Action of Anabaena Flos-Aquae Toxin. Science 1975, 187, 542–544. [Google Scholar]
- Rowell, R. R.; Wonnacott, S. Evidence for functional activity of up-regulated nicotine binding sites in rat striatal synaptosomes. J. Neurochem 1990, 55, 2105–2110. [Google Scholar]
- Stolerman, I. P.; Albuquerque, E. X.; Garcha, H. S. Behavioural effects of anatoxin, a potent nicotinic agonist, in rats. Neuropharmacology 1992, 31, 311–314. [Google Scholar]
- Lukas, R. J.; Changeux, J.-P.; le_Novere, N.; Albuquerque, E. X.; Balfour, D. J. K.; Berg, D. K.; Bertrand, D.; Chiappinelli, V. A.; Clarke, P. B. S.; Collins, A. C.; Dani, J. A.; Grady, S. A.; Kellar, K. J.; Lindstrom, J. M.; Marks, M. J.; Quik, M.; Taylor, P. W.; Wonnacott, S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol. Rev 1999, 51, 397–401. [Google Scholar]
- Thomas, P.; Stephens, M.; Wilkie, G.; Amar, M.; Lunt, G. G.; Whiting, P.; Gallagher, T.; Pereira, E.; Alkondon, M.; Albuquerque, E. X.; Wonnacott, S. (+)-Anatoxin-a is a potent agonist at neuronal nicotinic acetylcholine receptors. J. Neurochem. 1993, 60, 2308–2311. [Google Scholar]
- Jensen, A. A.; Frolund, B.; Liljefors, T.; Krogsgaard-Larsen, P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem 2005, 48, 4705–4745. [Google Scholar]
- MacAllan, D. R. E.; Lunt, G. G.; Wonnacott, S.; Aracava, Y.; Albuquerque, E. X. Methyllycaconitine and anatoxin-a differentiate between nicotinic receptors in vertebrate and invertebrate CNS. FEBS Lett 1988, 226, 357–363. [Google Scholar]
- Dajas-Bailador, F.; Wonnacott, S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci 2004, 25, 317–324. [Google Scholar]
- Cassels, B. K.; Bermudez, I.; Dajas, F.; Abin-Carriquiry, J. A.; Wonnacott, S. From ligand design to therapeutic efficacy: the challenge for nicotinic receptor research. Drug Discovery Today 2005, 10, 1657–1665. [Google Scholar]
- Devlin, J. P.; Edwards, O. E.; Gorham, P. R.; Hunter, N. R.; Pike, R. K.; Stavric, B. Anatoxin-a, a Toxic Alkaloid from Anabaena-Flos-Aquae Nrc-44h. Can. J. Chem 1977, 55, 1367–1371. [Google Scholar]
- Wonnacott, S.; Swanson, K. L.; Albuquerque, E. X.; Huby, N. J. S.; Thompson, P.; Gallagher, T. Homoanatoxin - a Potent Analog of Anatoxin-a. Biochem. Pharmacol 1992, 43, 419–423. [Google Scholar]
- Skulberg, O. M.; Carmichael, W. W.; Andersen, R. A.; Matsunaga, S.; Moore, R. E.; Skulberg, R. Investigations of a Neurotoxic Oscillatorialean Strain (Cyanophyceae) and Its Toxin - Isolation and Characterization of Homoanatoxin-A. Environ. Toxicol. Chem 1992, 11, 321–329. [Google Scholar]
- Zotou, A.; Jefferies, T. M.; Brough, P. A.; Gallagher, T. Determination of Anatoxin-a and Homoanatoxin in Blue-Green-Algal Extracts by High-Performance Liquid-Chromatography and Gas-Chromatography Mass-Spectrometry. Analyst 1993, 118, 753–758. [Google Scholar]
- Takada, N.; Iwatsuki, M.; Suenaga, K.; Uemura, D. Pinnamine, an alkaloidal marine toxin, isolated from Pinna muricata. Tetrahedron Lett 2000, 41, 6425–6428. [Google Scholar]
- Kigoshi, H.; Hayashi, N.; Uemura, D. Stereoselective synthesis of pinnamine, an alkaloidal marine toxin from Pinna muricata. Tetrahedron Lett 2001, 42, 7469–7471. [Google Scholar]
- Hjelmgaard, T.; Sotofte, I.; Tanner, D. Total synthesis of pinnamine and anatoxin-a via a common intermediate. A caveat on the anatoxin-a endgame. J. Org. Chem 2005, 70, 5688–5697. [Google Scholar]
- Schwarz, S.; Kampchen, T.; Tilotta, M. C.; Gundisch, D.; Seitz, G. Synthesis and nicotinic binding studies on enantiopure pinnamine variants with an 8-azabicyclo[3.2.1]octane moiety. Pharmazie 2003, 58, 295–299. [Google Scholar]
- Huber, C. S. The Crystal Structure and Absolute Configuration of 2,9-Diacetyl-9-azabicyclo[4,2,1]non-2,3-ene. Acta Crystallogr. Ser. B 1972, B28, 2577–2582. [Google Scholar]
- Namikoshi, M.; Murakami, T.; Fujiwara, T.; Nagai, H.; Niki, T.; Harigaya, E.; Watanabe, M. F.; Oda, T.; Yamada, J.; Tsujimura, S. Biosynthesis and transformation of homoanatoxin-a in the cyanobacterium Raphidiopsis mediterranea Skuja and structures of three new homologues. Chem. Res. Toxicol 2004, 17, 1692–1696. [Google Scholar]
- Hemscheidt, T.; Rapala, J.; Sivonen, K.; Skulberg, O. M. Biosynthesis of Anatoxin-a in Anabaena-Flos-Aquae and Homoanatoxin-a in Oscillatoria-Formosa. J. Chem. Soc., Chem. Commun. 1995, 1361–1362. [Google Scholar]
- Namikoshi, M.; Murakami, T.; Watanabe, M. F.; Oda, T.; Yamada, J.; Tsujimura, S.; Nagai, H.; Oishi, S. Simultaneous production of homoanatoxin-a, anatoxin-a, and a new non-toxic 4-hydroxyhomoanatoxin-a by the cyanobacterium Raphidiopsis mediterranea Skuja. Toxicon 2003, 42, 533–538. [Google Scholar]
- Gallon, J. R.; Chit, K. N.; Brown, E. G. Biosynthesis of the Tropane-Related Cyanobacterial Toxin Anatoxin-a - Role of Ornithine Decarboxylase. Phytochemistry 1990, 29, 1107–1111. [Google Scholar]
- Gallon, J. R.; Kittakoop, P.; Brown, E. G. Biosynthesis of Anatoxin-a by Anabaena-Flos-Aquae - Examination of Primary Enzymatic Steps. Phytochemistry 1994, 35, 1195–1203. [Google Scholar]
- Koskinen, A. M. P.; Rapoport, H. Synthetic and Conformational Studies on Anatoxin-a - a Potent Acetylcholine Agonist. J. Med. Chem 1985, 28, 1301–1309. [Google Scholar]
- Campbell, H. F.; Edwards, O. E.; Kolt, R. Synthesis of nor-Anatoxin-a and Anatoxin-a. Can. J. Chem 1977, 55, 1372–1379. [Google Scholar]
- Wegge, T.; Schwarz, S.; Seitz, G. A new and efficient synthetic route to enantiopure (+)-anatoxina from (−)-cocaine hydrochloride. Tetrahedron: Asymmetry 2000, 11, 1405–1410. [Google Scholar]
- Zhang, C. M.; Lomenzo, S. A.; Ballay, C. J.; Trudell, M. L. An improved synthesis of (+)-2-tropinone. J. Org. Chem 1997, 62, 7888–7889. [Google Scholar]
- Spivak, C. E.; Waters, J.; Witkop, B.; Albuquerque, E. X. Potencies and channel properties induced by semirigid agonists at frog nicotinic acetylcholine receptors. Mol. Pharmacol 1983, 23, 337–343. [Google Scholar]
- Badio, B.; Daly, J. W. Epibatidine, a potent analgesic and nicotinic agonist. Mol. Pharmacol 1994, 45, 563–569. [Google Scholar]
- Rozental, R.; Aracava, Y.; Scoble, G. T.; Swanson, K. L.; Wonnacott, S.; Albuquerque, E. X. Agonist recognition site of the peripheral acetylcholine receptor ion channel complex differentiates the enantiomers of nicotine. J. Pharmacol. Expt. Ther. 1989, 251, 395–404. [Google Scholar]
- Wonnacott, S. Alpha-Bungarotoxin binds to low-affinity nicotine binding sites in rat brain. J. Neurochem 1986, 47, 1706–1712. [Google Scholar]
- Beers, W. H.; Reich, E. Structure and Activity of Acetylcholine. Nature 1970, 228, 917–923. [Google Scholar]
- Sheridan, R. P.; Nilakantan, R.; Dixon, J. S.; Venkataraghavan, R. The Ensemble Approach to Distance Geometry - Application to the Nicotinic Pharmacophore. J. Med. Chem 1986, 29, 899– 906. [Google Scholar]
- Manallack, D.; Gallagher, T.; Livingstone, D. Principles in QSAR and Drug Design; Devillers, J., Ed.; Academic Press: London, 1996; Volume 2, pp. 177–208. [Google Scholar]
- Glennon, R. A.; Herndon, J. A.; Dukat, M. Epibatidine-Aided Studies toward Definition of a Nicotine Receptor Pharmacophore. Med. Chem. Res 1994, 6, 465–486. [Google Scholar]
- Tonder, J. E.; Olesen, P. H. Agonists at the alpha4beta2 nicotinic acetylcholine receptors: Structure-activity relationships and molecular modelling. Curr. Med. Chem 2001, 8, 651–674. [Google Scholar]
- Tonder, J. E.; Olesen, P. H.; Hansen, J. B.; Begtrup, M.; Pettersson, I. An improved nicotinic pharmacophore and a stereoselective CoMFA-model for nicotinic agonists acting at the central nicotinic acetylcholine receptors labelled by H-3-N-methylcarbamylcholine. J. Comput.-Aided. Mol. Des 2001, 15, 247–258. [Google Scholar]
- Tonder, J. E.; Hansen, J. B.; Begtrup, M.; Pettersson, I.; Rimvall, K.; Christensen, B.; Ehrbar, U.; Olesen, P. H. Improving the nicotinic pharmacophore with a series of (isoxazole)methylene-1-azacyclic compounds: Synthesis, structure-activity relationship, and molecular modeling. J. Med. Chem 1999, 42, 4970–4980. [Google Scholar]
- Glennon, R. A.; Dukat, M. alpha4beta2 nACh receptor pharmacophore models. Bioorg. Med. Chem. Lett 2004, 14, 1841–1844. [Google Scholar]
- Glennon, R. A.; Dukat, M.; Liao, L. Musings on alpha4beta2 nicotinic acetylcholine (nACh) receptor pharmacophore models. Curr. Med. Chem 2004, 4, 631–644. [Google Scholar]
- Manallack, D. T.; Ellis, D. D.; Thompson, P. E.; Gallagher, T.; Livingstone, D. J. Quantitative Structure-Activity Relationships of (+)-Anatoxin-a Derivatives. Natural Prod. Lett 1994, 4, 121–128. [Google Scholar]
- Petersen, J. S.; Fels, G.; Rapoport, H. Chirospecific Syntheses of (+)-Anatoxin and (−)-Anatoxin-a. J. Am. Chem. Soc 1984, 106, 4539–4547. [Google Scholar]
- Sardina, F. J.; Howard, M. H.; Morningstar, M.; Rapoport, H. Enantiodivergent Synthesis of (+)-Anatoxin and (−)-Anatoxin from L-Glutamic Acid. J. Org. Chem 1990, 55, 5025–5033. [Google Scholar]
- Sardina, F. J.; Howard, M. H.; Koskinen, A. M. P.; Rapoport, H. Chirospecific Synthesis of Nitrogen and Side-Chain Modified Analogs of (+)-Anatoxin. J. Org. Chem 1989, 54, 4654–4660. [Google Scholar]
- Howard, M. H.; Sardina, F. J.; Rapoport, H. Chirospecific Syntheses of Nitrogen and Side-Chain Modified Anatoxin Analogs - Synthesis of (1R)-Anatoxinal and (1R)-Anatoxinic Acid- Derivatives. J. Org. Chem 1990, 55, 2829–2838. [Google Scholar]
- Costa, A. C.; Swanson, K. L.; Aracava, Y.; Aronstam, R. S.; Albuquerque, E. X. Molecular effects of dimethylanatoxin on the peripheral nicotinic acetylcholine receptor. J. Pharmacol. Exp. Ther 1990, 252, 507–516. [Google Scholar]
- Kofuji, P.; Aracava, Y.; Swanson, K. L.; Aronstam, R. S.; Rapoport, H.; Albuquerque, E. X. Activation and blockade of the acetylcholine receptor-ion channel by the agonists (+)-anatoxin-a, the N-methyl derivative and the enantiomer. J. Pharmacol. Exp. Ther 1990, 252, 517–525. [Google Scholar]
- Wonnacott, S.; Jackman, S.; Swanson, K. L.; Rapoport, H.; Albuquerque, E. X. Nicotinic pharmacology of anatoxin analogs: II. Side chain structure-activity relationships at neuronal nicotinic ligand binding sites. J. Pharmacol. Exp. Ther 1991, 259, 387–391. [Google Scholar]
- Stevens, D. K.; Krieger, R. I. N-Methylation of anatoxin-a abolishes nicotinic cholinergic activity. Toxicon 1990, 28, 133–134. [Google Scholar]
- Swanson, K. L.; Aronstam, R. S.; Wonnacott, S.; Rapoport, H.; Albuquerque, E. X. Nicotinic pharmacology of anatoxin analogs. I. Side-chain structure-activity relationships at peripheral agonist and noncompetitive antagonist sites. J. Pharmacol. Exp. Ther 1991, 259, 377–386. [Google Scholar]
- Arneric, S. P.; Sullivan, J. P.; Briggs, C. A.; Donnelly-Roberts, D.; Anderson, D. J.; Raszkiewicz, J. L.; Hughes, M. L.; Cadman, E. D.; Adams, P.; Garvey, D. S.; et al. (S)-3-methyl-5-(1-methyl-2-pyrrolidinyl)isoxazole (ABT 418): a novel cholinergic ligand with cognition-enhancing and anxiolytic activities: I. In vitro characterization. J. Pharmacol. Exp. Ther 1994, 270, 310–318. [Google Scholar]
- Thomas, P.; Brough, P. A.; Gallagher, T.; Wonnacott, S. Alkyl-Modified Side-Chain Variants of Anatoxin-a - a Series of Potent Nicotinic Agonists. Drug Dev. Res 1994, 31, 147–156. [Google Scholar]
- Parsons, P. J.; Camp, N. P.; Edwards, N.; Sumoreeah, L. R. Synthesis of (+/−)-Anatoxin-a and Analogues. Tetrahedron 2000, 56, 309–315. [Google Scholar]
- Huby, N. J. S.; Thompson, P.; Wonnacott, S.; Gallagher, T. Structural Modification of Anatoxin-a - Synthesis of Model Affinity Ligands for the Nicotinic Acetylcholine-Receptor. J. Chem. Soc., Chem. Commun. 1991, 243–245. [Google Scholar]
- Magnus, N. A.; Ducry, L.; Rolland, V.; Wonnacott, S.; Gallagher, T. Direct C-11 functionalisation of anatoxin-a. Application to the synthesis of new ligand-based structural probes. J. Chem. Soc., Perkin Trans. 1 1997, 2313–2318. [Google Scholar]
- Thompson, P. E.; Manallack, D. T.; Blaney, F. E.; Gallagher, T. Conformational Studies on (+)- Anatoxin-a and Derivatives. J. Comput.-Aided Mol. Des 1992, 6, 287–298. [Google Scholar]
- Hacksell, U.; Mellin, C. Stereoselectivity of Nicotinic Receptors. Prog. Brain Res 1989, 79, 95–100. [Google Scholar]
- Kanne, D. B.; Ashworth, D. J.; Cheng, M. T.; Mutter, L. C.; Abood, L. G. Synthesis of the First Highly Potent Bridged Nicotinoid - 9-Azabicyclo[4.2.1]nona[2,3-c]pyridine (Pyrido[3,4- b]homotropane). J. Am. Chem. Soc 1986, 108, 7864–7865. [Google Scholar]
- Kanne, D. B.; Abood, L. G. Synthesis and Biological Characterization of Pyridohomotropanes - Structure-Activity-Relationships of Conformationally Restricted Nicotinoids. J. Med. Chem 1988, 31, 506–509. [Google Scholar]
- Gundisch, D.; Kampchen, T.; Schwarz, S.; Seitz, G.; Siegl, J.; Wegge, T. Syntheses and evaluation of pyridazine and pyrimidine containing bioisosteres of (+/−)-pyrido[3.4- b]homotropane and pyrido[3.4-b]tropane as novel nAChR ligands. Bioorg. Med. Chem 2002, 10, 1–9. [Google Scholar]
- Brough, P. A.; Gallagher, T.; Thomas, P.; Wonnacott, S.; Baker, R.; Malik, K. M. A.; Hursthouse, M. B. Synthesis and X-Ray Crystal-Structure of 2-Acetyl-9-Azabicyclo[4.2.1]nonan-3-one - a Conformationally Locked s-Cis Analog of Anatoxin-a. J. Chem. Soc., Chem. Commun. 1992, 1087–1089. [Google Scholar]
- Hernandez, A.; Rapoport, H. Conformationally Constrained Analogs of Anatoxin - Chirospecific Synthesis of s-Trans Carbonyl Ring-Fused Analogs. J. Org. Chem 1994, 59, 1058–1066. [Google Scholar]
- Holladay, M. W.; Dart, M. J.; Lynch, J. K. Neuronal nicotinic acetylcholine receptors as targets for drug discovery. J. Med. Chem 1997, 40, 4169–4194. [Google Scholar]
- Le Novere, N.; Grutter, T.; Changeux, J. P. Models of the extracellular domain of the nicotinic receptors and of agonist- and Ca2+-binding sites. Proc. Natl. Acad. Sci. U S A 2002, 99, 3210–3215. [Google Scholar]
- Wright, E.; Gallagher, T.; Sharples, C. G. V.; Wonnacott, S. Synthesis of UB-165: A novel nicotinic ligand and anatoxin-a/epibatidine hybrid. Bioorg. Med. Chem. Lett 1997, 7, 2867–2870. [Google Scholar]
- Sharples, C. G.; Kaiser, S.; Soliakov, L.; Marks, M. J.; Collins, A. C.; Washburn, M.; Wright, E.; Spencer, J. A.; Gallagher, T.; Whiteaker, P.; Wonnacott, S. UB-165: a novel nicotinic agonist with subtype selectivity implicates the alpha4beta2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J. Neurosci 2000, 20, 2783–2791. [Google Scholar]
- Cao, Y. J.; Surowy, C. S.; Puttfarcken, P. S. Nicotinic acetylcholine receptor-mediated [3H]dopamine release from hippocampus. J. Pharmacol. Exp. Ther 2005, 312, 1298–1304. [Google Scholar]
- Sharples, C. G. V.; Karig, G.; Simpson, G. L.; Spencer, J. A.; Wright, E.; Millar, N. S.; Wonnacott, S.; Gallagher, T. Synthesis and pharmacological characterization of novel analogues of the nicotinic acetylcholine receptor agonist (+/−)-UB-165. J. Med. Chem 2002, 45, 3235–3245. [Google Scholar]
- Gohlke, H.; Gundisch, D.; Schwarz, S.; Seitz, G.; Tilotta, M. C.; Wegge, T. Synthesis and nicotinic binding studies on enantiopure diazine analogues of the novel (2-chloro-5-pyridyl)-9- azabicyclo[4.2.1]non-2-ene UB-165. J. Med. Chem 2002, 45, 1064–1072. [Google Scholar]
- Karig, G.; Large, J. M.; Sharples, C. G. V.; Sutherland, A.; Gallagher, T.; Wonnacott, S. Synthesis and nicotinic binding of novel phenyl derivatives of UB-165. Identifying factors associated with alpha7 selectivity. Bioorg. Med. Chem. Lett 2003, 13, 2825–2828. [Google Scholar]
- Sutherland, A.; Gallagher, T.; Sharples, C. G. V.; Wonnacott, S. Synthesis of two fluoro analogues of the nicotinic acetylcholine receptor agonist UB-165. J. Org. Chem 2003, 68, 2475–2478. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

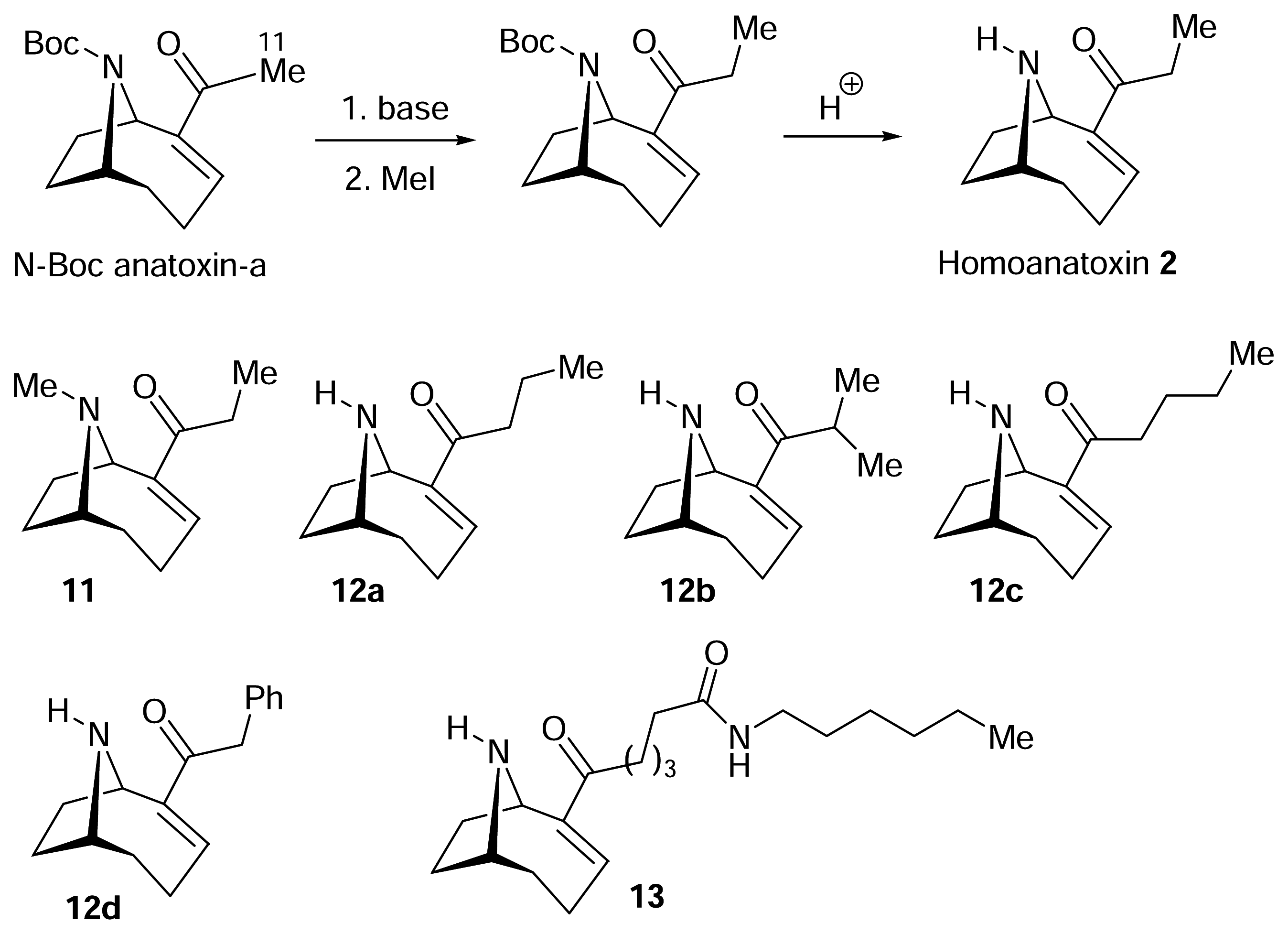
 ) [50–53].
) [50–53].









© 2006 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Wonnacott, S.; Gallagher, T. The Chemistry and Pharmacology of Anatoxin-a and Related Homotropanes with respect to Nicotinic Acetylcholine Receptors. Mar. Drugs 2006, 4, 228-254. https://doi.org/10.3390/md403228
Wonnacott S, Gallagher T. The Chemistry and Pharmacology of Anatoxin-a and Related Homotropanes with respect to Nicotinic Acetylcholine Receptors. Marine Drugs. 2006; 4(3):228-254. https://doi.org/10.3390/md403228
Chicago/Turabian StyleWonnacott, Susan, and Timothy Gallagher. 2006. "The Chemistry and Pharmacology of Anatoxin-a and Related Homotropanes with respect to Nicotinic Acetylcholine Receptors" Marine Drugs 4, no. 3: 228-254. https://doi.org/10.3390/md403228
APA StyleWonnacott, S., & Gallagher, T. (2006). The Chemistry and Pharmacology of Anatoxin-a and Related Homotropanes with respect to Nicotinic Acetylcholine Receptors. Marine Drugs, 4(3), 228-254. https://doi.org/10.3390/md403228