
Endocrine Aspects of Environmental “Obesogen” Pollutants

 ,
,  ,
, 

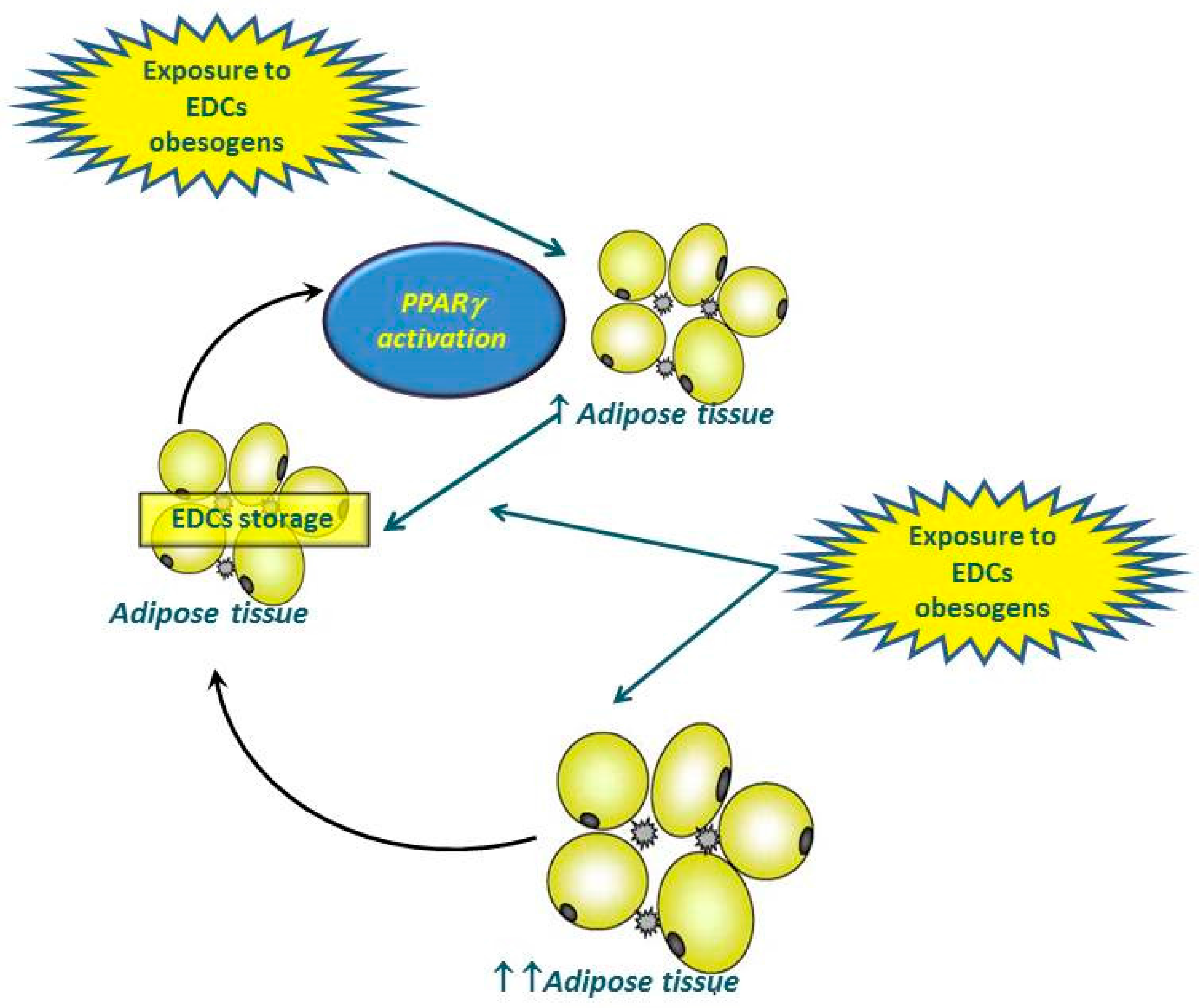{kind=link}
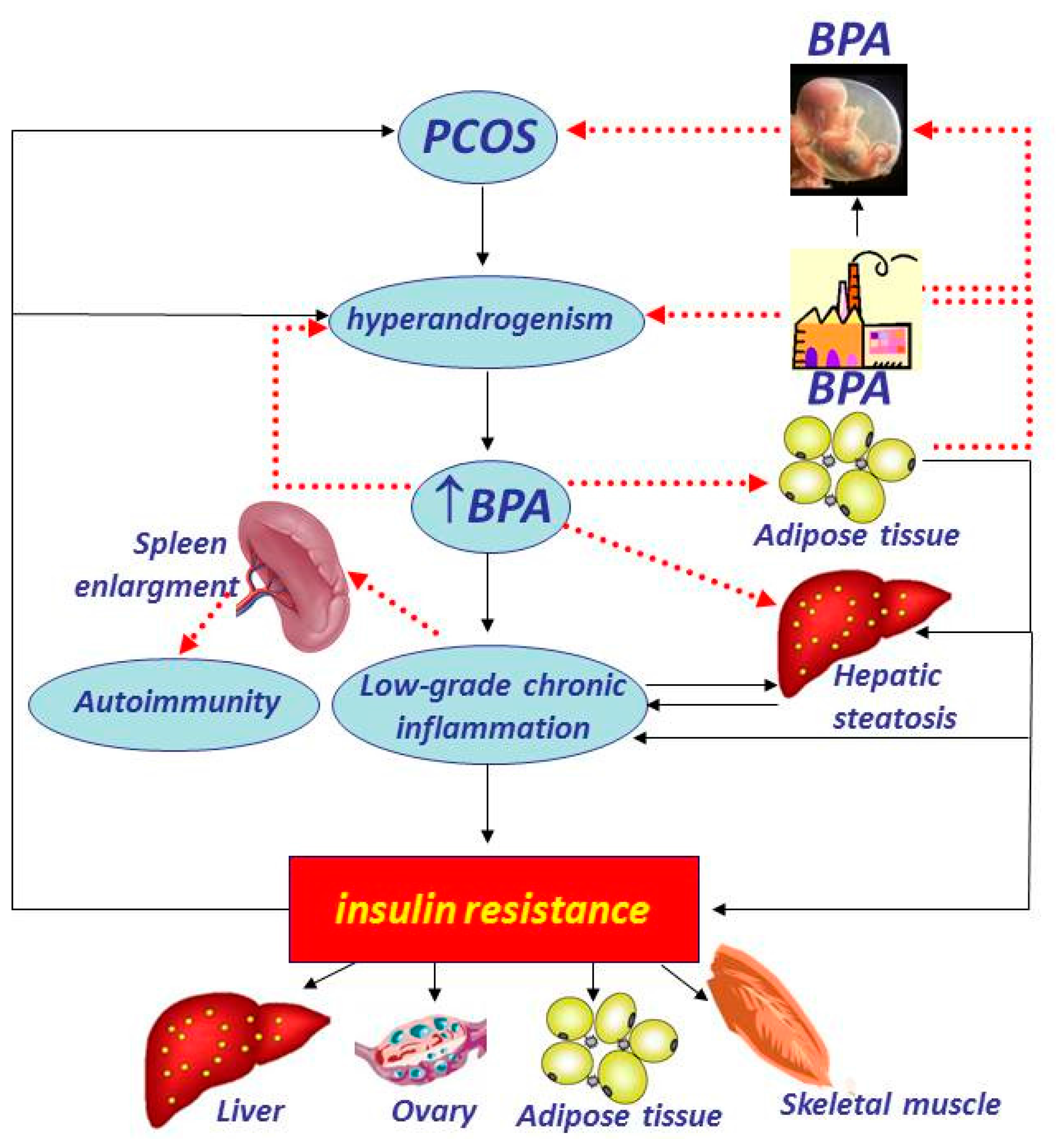{kind=link}
Abstract
:1. Introduction
2. Endocrine-Disrupting Chemicals (EDCs)
3. “Obesogenic” EDCs from the Endocrinologic Point of View
4. Diethylstilbestrol
5. Bisphenol A
6. Phytoestrogens
7. Phthalates
8. Organochlorine and Organophosphate Pesticides
9. Polychlorinated Bisphenols
10. Perfluoroalkyl Substances
11. Inhaled Pollutants
12. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Swinburn, B.; Egger, G. Preventive strategies against weight gain and obesity. Obes. Rev. 2002, 3, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, S.; O’Rahilly, S. Genetics of obesity in humans. Endocr. Rev. 2006, 27, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Pigeyre, M.; Yazdi, F.T.; Kaur, Y.; Meyre, D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin. Sci. (Lond.) 2016, 130, 943–986. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, M.; Ismail, K.; Gervin, K.; Kyllönen, A.; Hakkarainen, A.; Lundbom, J.; Järvinen, E.A.; Harris, J.R.; Lundbom, N.; Rissanen, A.; et al. Genome-wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin. Epigenet. 2015, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stel, J.; Legler, J. The Role of Epigenetics in the Latent Effects of Early Life Exposure to Obesogenic Endocrine Disrupting Chemicals. Endocrinology 2015, 156, 3466–3472. [Google Scholar] [CrossRef] [PubMed]
- Dubowsky, S.D.; Suh, H.; Schwartz, J.; Coull, B.A.; Gold, D.R. Diabetes, obesity, and hypertension may enhance associations between air pollution and markers of systemic inflammation. Environ. Health Perspect. 2006, 114, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s second scientific statement on endocrine-disrupting chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef] [PubMed]
- Rijk, I.; van Duursen, M.; van den Berg, M. Health Cost that May be Associated with Endocrine Disrupting Chemicals; Universiteit Utrecht, Institute for Risk Assessment Sciences: Utrecht, The Netherlands, 2016. [Google Scholar]
- World Health Organization (WHO). State of the Science of Endocrine Disrupting Chemicals—2012. Available online: http://www.who.int/ceh/publications/endocrine/en/ (accessed on 27 July 2016).
- Vandenberg, L.N.; Colborn, T.; Hayes, T.B.; Heindel, J.J.; Jacobs, D.R., Jr.; Lee, D.H.; Shioda, T.; Soto, A.M.; vom Saal, F.S.; Welshons, W.V.; et al. Hormones and Endocrine-Disrupting Chemicals: Low-Dose Effects and Nonmonotonic Dose Responses. Endocr. Rev. 2012, 33, 378–455. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, F.; Mostafalou, S.; Bahadar, H.; Abdollahi, M. Review of endocrine disorders associated with environmental toxicants and possible involved mechanisms. Life Sci. 2016, 145, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.S.; Blumberg, B. Obesogens: An emerging threat to public health. Am. J. Obstet. Gynecol. 2016, 214, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Baillie-Hamilton, P.F. Chemical toxins: A hypothesis to explain the global obesity epidemic. J. Altern. Complement. Med. 2002, 8, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Riu, A.; McCollum, C.W.; Pinto, C.L.; Grimaldi, M.; Hillenweck, A.; Perdu, E.; Zalko, D.; Bernard, L.; Laudet, V.; Balaguer, P.; et al. Halogenated Bisphenol-A analogs act as obesogens in Zebrafish larvae (Danio rerio). Toxicol. Sci. 2014, 139, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Strakovsky, R.S.; Lezmi, S.; Flaws, J.A.; Schantz, S.L.; Pan, Y.X.; Helferich, W.G. Genistein exposure during the early postnatal period favors the development of obesity in female, but not male rats. Toxicol. Sci. 2014, 138, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Yue, P.; Deiuliis, J.A.; Lumeng, C.N.; Kampfrath, T.; Mikolaj, M.B.; Cai, Y.; Ostrowski, M.C.; Lu, B.; Parthasarathy, S.; et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation 2009, 119, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Grün, F.; Blumberg, B. Environmental obesogens: Organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 2006, 147 (Suppl. 6), S50–S55. [Google Scholar] [CrossRef] [PubMed]
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—An allostatic perspective. Biochim. Biophys. Acta 2010, 1801, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Darbre, P.D. Endocrine disruption and disorders of energy metabolism. In Endocrine Disruptors and Human Health; Darbre, P.D., Ed.; Elsevier: London, UK, 2015; pp. 273–289. [Google Scholar]
- Mostafalou, S. Persistent organic pollutants and concern over the link with insulin resistance related metabolic diseases. Rev. Environ. Contam. Toxicol. 2016, 238, 69–89. [Google Scholar] [PubMed]
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Capone, D.; Finelli, C. Exposure to ambient air particulate matter and non-alcoholic fatty liver disease. World J. Gastroenterol. 2013, 19, 3951–3956. [Google Scholar] [CrossRef] [PubMed]
- Minihane, A.M.; Vinoy, S.; Russell, W.R.; Baka, A.; Roche, H.M.; Tuohy, K.M.; Teeling, J.L.; Blaak, E.E.; Fenech, M.; Vauzour, D.; et al. Low-grade inflammation, diet composition and health: Current research evidence and its translation. Br. J. Nutr. 2015, 114, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; vom Saal, F.S.; Blumberg, B.; Bovolin, P.; Calamandrei, G.; Ceresini, G.; Cohn, B.A.; Fabbri, E.; Gioiosa, L.; Kassotis, C.; et al. Parma consensus statement on metabolic disruptors. Environ. Health 2015, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Deretzi, G.; Zavos, C.; Mantzoros, C.S. The emerging role of endocrine disruptors in pathogenesis of insulin resistance: A concept implicating nonalcoholic fatty liver disease. Curr. Mol. Med. 2012, 12, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.M.; Johnson, A.; Tarapore, P.; Janakiram, V.; Zhang, X.; Leung, Y.K. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J. 2012, 53, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Cheikh Rouhou, M.; Karelis, A.D.; St-Pierre, D.H.; Lamontagne, L. Adverse effects of weight loss: Are persistent organic pollutants a potential culprit? Diabetes Metab. 2016, S1262–S3636, 30418–30419. [Google Scholar] [CrossRef] [PubMed]
- Guénard, F.; Tchernof, A.; Deshaies, Y.; Cianflone, K.; Kral, J.G.; Marceau, P.; Vohl, M.C. Methylation and expression of immune and inflammatory genes in the offspring of bariatric bypass surgery patients. J. Obes. 2013, 2013, 492170. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; Molloy, P.L.; Varinli, H.; Morrison, J.L.; Muhlhausler, B.S. Members of EpiSCOPE epigenetics and human obesity. Int. J. Obes. (Lond.) 2015, 39, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Janani, C.; Ranjitha Kumari, B.D. PPAR gamma gene—A review. Diabetes Metab. Syndr. 2015, 9, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Snedeker, S.M.; Hay, A.G. Do interactions between gut ecology and environmental chemicals contribute to obesity and diabetes? Environ. Health Perspect. 2012, 120, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Nichols, R.G.; Correll, J.; Murray, I.A.; Tanaka, N.; Smith, P.B.; Hubbard, T.D.; Sebastian, A.; Albert, I.; Hatzakis, E.; et al. Persistent organic pollutants modify gut microbiota-host metabolic homeostasis in mice through aryl hydrocarbon receptor activation. Environ. Health Perspect. 2015, 123, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, I.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor and colitis. Semin. Immunopathol. 2013, 35, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Potera, C. POPs and gut microbiota: Dietary exposure alters ratio of bacterial species. Environ. Health Perspect. 2015, 123, A187. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.E.; Troisi, R.; Palmer, J.R.; Wise, L.A.; Titus, L.; Strohsnitter, W.C.; Ricker, W.; Hyer, M.; Hoover, R.N. Prenatal diethylstilbestrol exposure and risk of obesity in adult women. J. Dev. Orig. Health Dis. 2015, 6, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Hoover, R.N.; Hyer, M.; Pfeiffer, R.M.; Adam, E.; Bond, B.; Cheville, A.L.; Colton, T.; Hartge, P.; Hatch, E.E.; Herbst, A.L.; et al. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N. Engl. J. Med. 2011, 365, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Newbold, R.R.; Padilla-Banks, E.; Snyder, R.J.; Phillips, T.M.; Jefferson, W.N. Developmental exposure to endocrine disruptors and the obesity epidemic. Reprod. Toxicol. 2007, 23, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Newbold, R.R. Developmental exposure to endocrine-disrupting chemicals programs for reproductive tract alterations and obesity later in life. Am. J. Clin. Nutr. 2011, 94 (Suppl. 6), 1939S–1942S. [Google Scholar] [CrossRef] [PubMed]
- Edlow, A.G.; Chen, M.; Smith, N.A.; Lu, C.; McElrath, T.F. Fetal bisphenol A exposure: Concentration of conjugated and unconjugated Bisphenol A in amniotic fluid in the second and third trimesters. Reprod. Toxicol. 2012, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ikezuki, Y.; Tsutsumi, O.; Takai, Y.; Kamei, Y.; Taketani, Y. Determination of Bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum. Reprod. 2002, 17, 2839–2841. [Google Scholar] [CrossRef] [PubMed]
- Deceuninck, Y.; Bichon, E.; Marchand, P.; Boquien, C.Y.; Legrand, A.; Boscher, C.; Antignac, J.P.; Le Bizec, B. Determination of Bisphenol A and related substitutes/analogues in human breast milk using gas chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.A.; Birnbaum, L.S.; Farabollini, F.; Newbold, R.R.; Rubin, B.S.; Talsness, C.E.; Vandenbergh, J.G.; Walser-Kuntz, D.R.; vom Saal, F.S. In vivo effects of Bisphenol A in laboratory rodent studies. Reprod. Toxicol. 2007, 24, 199–224. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.H.; Pallaroni, L.; Yoon, K.; Gaido, K.; Ross, S.; McDonnell, D. Problems for risk assessment of endocrine-active estrogenic compounds. Environ. Health Perspect. 2002, 110 (Suppl. 6), 925–929. [Google Scholar] [CrossRef] [PubMed]
- Hayes, L.; Weening, A.; Morey, L.M. Differential effects of estradiol and Bisphenol A on SET8 and SIRT1 expression in ovarian cancer cells. Dose Response 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Chen, D.; He, Y.; Zhu, W.; Zhou, W.; Zhang, J. Bisphenol-A and female infertility: A possible role of gene-environment interactions. Int. J. Environ. Res. Public Health 2015, 12, 11101–11116. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, C.; Tait, S.; Guerranti, C.; Busani, L.; Ciardo, F.; Bergamasco, B.; Perra, G.; Mancini, F.R.; Marci, R.; Bordi, G.; et al. Exposure to endocrine disruptors and nuclear receptors gene expression in infertile and fertile men from Italian areas with different environmental features. Int. J. Environ. Res. Public Health 2015, 12, 12426–12445. [Google Scholar] [CrossRef] [PubMed]
- Ben-Jonathan, N.; Hugo, E.R.; Brandebourg, T.D. Effects of Bisphenol A on adipokine release from human adipose tissue: Implications for the metabolic syndrome. Mol. Cell Endocrinol. 2009, 304, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, N.; Fénichel, P. Endocrine disruptors: New players in the pathophysiology of type 2 diabetes? Diabetes Metab. 2015, 41, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Carwile, J.L.; Karin, B.; Michels, K.B. Urinary Bisphenol A and obesity: NHANES 2003–2006. Environ. Res. 2011, 111, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.; D’Esposito, V.; Passaretti, F.; Liotti, A.; Cabaro, S.; Longo, M.; Perruolo, G.; Oriente, F.; Beguinot, F.; Formisano, P. Bisphenol-A impairs insulin action and up-regulates inflammatory pathways in human subcutaneous adipocytes and 3T3-L1 cells. PLoS ONE 2013, 8, e82099. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Cortese, K.; Voci, A.; Vergani, L.; Fabbri, R.; Barmo, C.; Gallo, G.; Canesi, L. Direct effects of Bisphenol A on lipid homeostasis in rat hepatoma cells. Chemosphere 2013, 91, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Ariemma, F.; D’Esposito, V.; Liguoro, D.; Oriente, F.; Cabaro, S.; Liotti, A.; Cimmino, I.; Longo, M.; Beguinot, F.; Formisano, P.; et al. Low-dose Bisphenol-A impairs adipogenesis and generates dysfunctional 3T3-L1 adipocytes. PLoS ONE 2016, 11, e0150762. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, J.F.; Strong, A.L.; McLachlan, J.A.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Bisphenol A enhances adipogenic differentiation of human adipose stromal/stem cells. J. Mol. Endocrinol. 2014, 53, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, B.; Hou, M.; Pan, X.; Li, X. The environmental obesogen Bisphenol A promotes adipogenesis by increasing the amount of 11β-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int. J. Obes. (Lond.) 2013, 37, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.G.; Boudreau, A.; Atlas, E. Bisphenol A induces differentiation of human preadipocytes in the absence of glucocorticoid and is inhibited by an estrogen-receptor antagonist. Nutr. Diabetes 2014, 4, e102. [Google Scholar] [CrossRef] [PubMed]
- Savastano, S.; Tarantino, G.; D’Esposito, V.; Passaretti, F.; Cabaro, S.; Liotti, A.; Liguoro, D.; Perruolo, G.; Ariemma, F.; Finelli, C.; et al. Bisphenol-A plasma levels are related to inflammatory markers, visceral obesity and insulin-resistance: A cross-sectional study on adult male population. J. Transl. Med. 2015, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Valentino, R.; Di Somma, C.; D’Esposito, V.; Passaretti, F.; Pizza, G.; Brancato, V.; Orio, F.; Formisano, P.; Colao, A.; et al. Bisphenol A in polycystic ovary syndrome and its association with liver-spleen axis. Clin. Endocrinol. (Oxf.) 2013, 78, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Behloul, N.; Wu, G. Genistein: A promising therapeutic agent for obesity and diabetes treatment. Eur. J. Pharmacol. 2013, 698, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Patisaul, H.B.; Jefferson, W. The pros and cons of phytoestrogens. Front. Neuroendocrinol. 2010, 31, 400–419. [Google Scholar] [CrossRef] [PubMed]
- Penza, M.; Montani, C.; Romani, A.; Vignolini, P.; Pampaloni, B.; Tanini, A.; Brandi, M.L.; Alonso-Magdalena, P.; Nadal, A.; Ottobrini, L.; et al. Genistein affects adipose tissue deposition in a dose-dependent and gender-specific manner. Endocrinology 2006, 147, 5740–5751. [Google Scholar] [CrossRef] [PubMed]
- Calafat, A.M.; McKee, R.H. Integrating biomonitoring exposure data into the risk assessment process: Phthalates [diethyl phthalate and di(2-ethylhexyl) phthalate] as a case study. Environ. Health Perspect. 2006, 114, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, M.J. Phthalate exposure and childhood obesity. Ann. Pediatr. Endocrinol. Metab. 2014, 19, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.E.; Nelson, J.W.; Qureshi, M.M.; Weinberg, J.; Moore, L.L.; Singer, M.; Webster, T.F. Association of urinary phthalate metabolite concentrations with body mass index and waist circumference: A cross-sectional study of NHANES data, 1999–2002. Environ. Health 2008, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Buser, M.C.; Murray, H.E.; Scinicariello, F. Age and sex differences in childhood and adulthood obesity association with phthalates: Analyses of NHANES 2007–2010. Int. J. Hyg. Environ. Health 2014, 217, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.M.; Jones, T.H. Testosterone and obesity. Obes. Rev. 2015, 16, 581–606. [Google Scholar] [CrossRef] [PubMed]
- Svechnikov, K.; Izzo, G.; Landreh, L.; Weisser, J.; Söder, O. Endocrine disruptors and Leydig cell function. J. Biomed. Biotechnol. 2010, 2010, 684504. [Google Scholar] [CrossRef] [PubMed]
- Rouiller-Fabre, V.; Guerquin, M.J.; N’Tumba-Byn, T.; Muczynski, V.; Moison, D.; Tourpin, S.; Messiaen, S.; Habert, R.; Livera, G. Nuclear Receptors and Endocrine Disruptors in Fetal and Neonatal Testes: A Gapped Landscape. Front. Endocrinol. (Lausanne) 2015, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A. Does early-life exposure to organophosphate insecticides lead to prediabetes and obesity? Reprod. Toxicol. 2011, 31, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Steffes, M.W.; Sjödin, A.; Jones, R.S.; Needham, L.L.; Jacobs, D.R., Jr. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS ONE 2011, 6, e15977. [Google Scholar] [CrossRef] [PubMed]
- Orton, F.; Rosivatz, E.; Scholze, M.; Kortenkamp, A. Widely used pesticides with previously unknown endocrine activity revealed as in vitro antiandrogens. Environ. Health Perspect. 2011, 119, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Mangum, L.H.; Howell, G.E.; Chambers, J.E. Exposure to p,p’-DDE enhances differentiation of 3T3-L1 preadipocytes in a model of sub-optimal differentiation. Toxicol. Lett. 2015, 238, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Peris-Sampedro, F.; Basaure, P.; Reverte, I.; Cabré, M.; Domingo, J.L.; Colomina, M.T. Chronic exposure to chlorpyrifos triggered body weight increase and memory impairment depending on human apoE polymorphisms in a targeted replacement mouse model. Physiol. Behav. 2015, 144, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Murinova, L.; Trnovec, T.; Loffredo, C.A.; Washington, K.; Mitra, P.S.; Dutta, S.K. Biomarkers linking PCB exposure and obesity. Curr. Pharm. Biotechnol. 2014, 15, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Portigal, C.L.; Cowell, S.P.; Fedoruk, M.N.; Butler, C.M.; Rennie, P.S.; Nelson, C.C. Polychlorinated biphenyls interfere with androgen-induced transcriptional activation and hormone binding. Toxicol. Appl. Pharmacol. 2002, 179, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Zota, A.R.; Park, J.S.; Wang, Y.; Petreas, M.; Zoeller, R.T.; Woodruff, T.J. Polybrominated diphenyl ethers, hydroxylated polybrominated diphenyl ethers, and measures of thyroid function in second trimester pregnant women in California. Environ. Sci. Technol. 2011, 45, 7896–7905. [Google Scholar] [CrossRef] [PubMed]
- Tang-Péronard, J.L.; Heitmann, B.L.; Andersen, H.R.; Steuerwald, U.; Grandjean, P.; Weihe, P.; Jensen, T.K. Association between prenatal polychlorinated biphenyl exposure and obesity development at ages 5 and 7 y: A prospective cohort study of 656 children from the Faroe Islands. Am. J. Clin. Nutr. 2014, 99, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Lignell, S.; Aune, M.; Darnerud, P.O.; Hanberg, A.; Larsson, S.C.; Glynn, A. Prenatal exposure to polychlorinated biphenyls (PCBs) and polybrominated diphenyl ethers (PBDEs) may influence birth weight among infants in a Swedish cohort with background exposure: A cross-sectional study. Environ. Health 2013, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Valvi, D.; Mendez, M.A.; Martinez, D.; Grimalt, J.O.; Torrent, M.; Sunyer, J.; Vrijheid, M. Prenatal concentrations of polychlorinated biphenyls, DDE, and DDT and overweight in children: A prospective birth cohort study. Environ. Health Perspect. 2012, 120, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Buck, R.C.; Franklin, J.; Berger, U.; Conder, J.M.; Cousins, I.T.; de Voogt, P.; Jensen, A.A.; Kannan, K.; Mabury, S.A.; van Leeuwen, S.P. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integr. Environ. Assess. Manag. 2011, 7, 513–541. [Google Scholar] [CrossRef] [PubMed]
- D’Hollander, W.; de Voogt, P.; De Coen, W.; Bervoets, L. Perfluorinated substances in human food and other sources of human exposure. Rev. Environ. Contam. Toxicol. 2010, 208, 179–215. [Google Scholar] [PubMed]
- Braun, J.M.; Chen, A.; Romano, M.E.; Calafat, A.M.; Webster, G.M.; Yolton, K.; Lanphear, B.P. Prenatal perfluoroalkyl substance exposure and child adiposity at 8 years of age: The HOME study. Obesity (Silver Spring) 2016, 24, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.M.; Oken, E.; Rifas-Shiman, S.L.; Webster, T.F.; Gillman, M.W.; Calafat, A.M.; Ye, X.; Sagiv, S.K. Prenatal Exposure to Perfluoroalkyl Substances and Adiposity in Early and Mid-Childhood. Environ. Health Perspect. 2016, 28. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Zhang, H.; Zheng, F.; Sheng, N.; Guo, X.; Dai, J. Perfluorooctanoic acid exposure for 28 days affects glucose homeostasis and induces insulin hypersensitivity in mice. Sci. Rep. 2015, 5, 11029. [Google Scholar] [CrossRef] [PubMed]
- Starling, A.P.; Engel, S.M.; Richardson, D.B.; Baird, D.D.; Haug, L.S.; Stuebe, A.M.; Klungsøyr, K.; Harmon, Q.; Becher, G.; Thomsen, C.; et al. Perfluoroalkyl substances during pregnancy and validated preeclampsia among nulliparous women in the Norwegian Mother and Child Cohort Study. Am. J. Epidemiol. 2014, 179, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Rajagopalan, S.; Pope CA, I.I.I.; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease an update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef] [PubMed]
- Weichenthal, S.; Hoppin, J.A.; Reeves, F. Obesity and the cardiovascular health effects of fine particulate air pollution. Obesity (Silver Spring) 2014, 22, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Bind, M.A.; Koutrakis, P.; Coull, B.A.; Sparrow, D.; Vokonas, P.S.; Schwartz, J.D. Fine particles, genetic pathways, and markers of inflammation and endothelial dysfunction: Analysis on particulate species and sources. J. Expo. Sci. Environ. Epidemiol. 2016, 26, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yavar, Z.; Verdin, M.; Ying, Z.; Mihai, G.; Kampfrath, T.; Wang, A.; Zhong, M.; Lippmann, M.; Chen, L.C.; et al. Effect of early particulate air pollution exposure on obesity in mice: Role of p47phox. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Hiraiwa, K.; van Eeden, S.F. Contribution of Lung Macrophages to the Inflammatory Responses Induced by Exposure to Air Pollutants. Mediat. Inflamm. 2013, 2013, 619523. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, R.; Rocic, P. The metabolic syndrome, oxidative stress, environment, and cardiovascular disease: The great exploration. Exp. Diabetes Res. 2012, 2012, 271028. [Google Scholar] [CrossRef] [PubMed]
- Eze, I.C.; Schaffner, E.; Foraster, M.; Imboden, M.; von Eckardstein, A.; Gerbase, M.W.; Rothe, T.; Rochat, T.; Künzli, N.; Schindler, C.; et al. Long-term exposure to ambient air pollution and metabolic syndrome in adults. PLoS ONE 2015, 10, e0130337. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.H.; Fiel, M.I.; Sun, Q.; Guo, J.; Gordon, R.E.; Chen, L.C.; Friedman, S.L.; Odin, J.A.; Allina, J. Kupffer cell activation by ambient air particulate matter exposure may exacerbate non-alcoholic fatty liver disease. J. Immunotoxicol. 2009, 6, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.M.; Pal, S.; Guénette, J.; Wade, M.G.; Atlas, E.; Holloway, A.C.; Williams, A.; Vincent, R. Ozone inhalation provokes glucocorticoid-dependent and -independent effects on inflammatory and metabolic pathways. Toxicol. Sci. 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Ferraù, F.; Korbonits, M. Metabolic comorbidities in Cushing’s syndrome. Eur. J. Endocrinol. 2015, 173, M133–M157. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.; McQueen, A.; Chen, T.C.; Wang, J.C. Regulation of glucose homeostasis by glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar] [PubMed]
- Sominsky, L.; Spencer, S.J. Eating behavior and stress: A pathway to obesity. Front. Psychol. 2014, 5, 434. [Google Scholar] [CrossRef] [PubMed]
- Björntorp, P.; Rosmond, R. Obesity and cortisol. Nutrition 2000, 16, 924–936. [Google Scholar] [CrossRef]
- Harris, R.B. Chronic and acute effects of stress on energy balance: Are there appropriate animal models? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R250–R265. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.; Heindel, J.J.; Kasten, T.; Kidd, K.A.; Jobling, S.; Neira, M.; Zoeller, R.T.; Becher, G.; Bjerreqaard, P.; Bornman, R.; et al. The impact of endocrine disruption: A consensus statement on the state of the science Editorial Bergman A. Environ. Health Perspect. 2013, 121, A104–A106. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F.; Barrea, L.; Di Somma, C.; Savanelli, M.C.; Muscogiuri, G.; Orio, F.; Savastano, S. Endocrine Aspects of Environmental “Obesogen” Pollutants. Int. J. Environ. Res. Public Health 2016, 13, 765. https://doi.org/10.3390/ijerph13080765
Nappi F, Barrea L, Di Somma C, Savanelli MC, Muscogiuri G, Orio F, Savastano S. Endocrine Aspects of Environmental “Obesogen” Pollutants. International Journal of Environmental Research and Public Health. 2016; 13(8):765. https://doi.org/10.3390/ijerph13080765
Chicago/Turabian StyleNappi, Francesca, Luigi Barrea, Carolina Di Somma, Maria Cristina Savanelli, Giovanna Muscogiuri, Francesco Orio, and Silvia Savastano. 2016. "Endocrine Aspects of Environmental “Obesogen” Pollutants" International Journal of Environmental Research and Public Health 13, no. 8: 765. https://doi.org/10.3390/ijerph13080765
APA StyleNappi, F., Barrea, L., Di Somma, C., Savanelli, M. C., Muscogiuri, G., Orio, F., & Savastano, S. (2016). Endocrine Aspects of Environmental “Obesogen” Pollutants. International Journal of Environmental Research and Public Health, 13(8), 765. https://doi.org/10.3390/ijerph13080765