In Vitro Assessment of the Efficacy of a Macrocyclic Chelator in Reversing Methylmercury Toxicity




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Determination of TrxR Activity by the DTNB Reduction Assay
2.3. Trx System Activity Determination by Insulin Reduction Assay
2.4. Cell Culture
2.5. Cell Viability Assay
2.6. Preparation of Cell Lysates
2.7. Total Soluble Protein
2.8. TrxR and Trx Activity Determination
2.9. Expression of TrxR and Trx
2.10. Statistical Analysis
3. Results
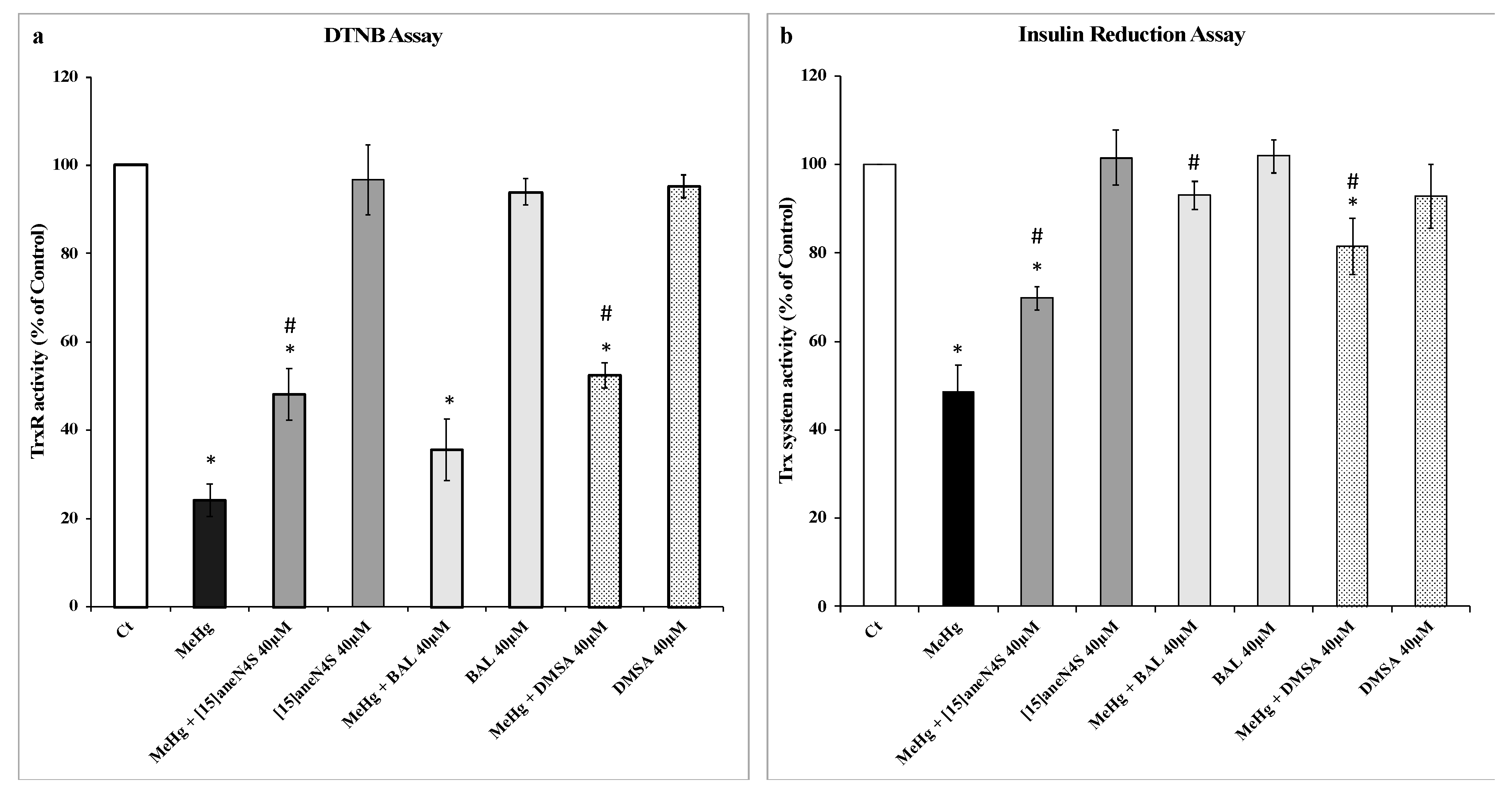
3.1. Effect of MeHg and Chelating Agents on TrxR and Trx Activities
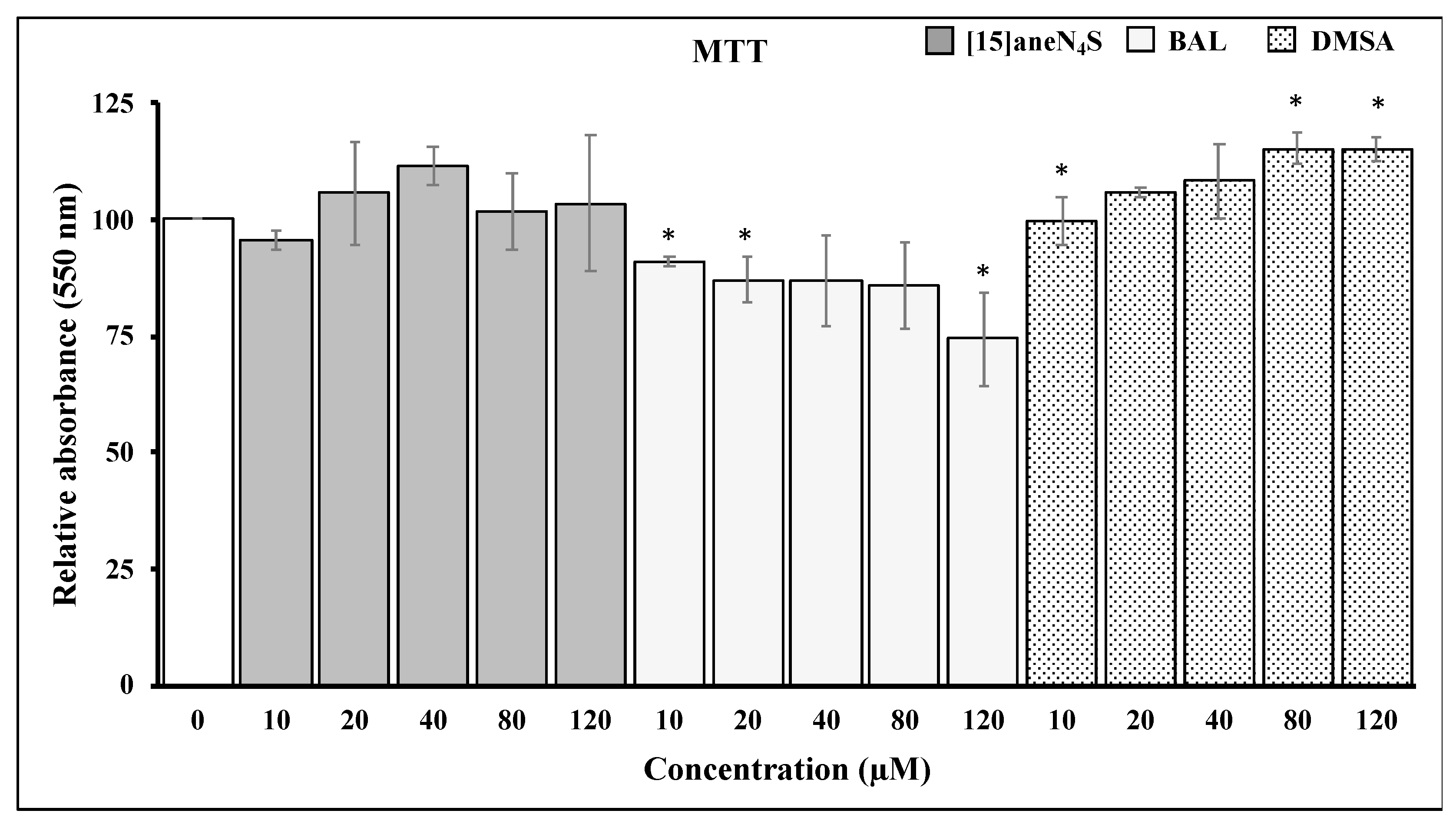
3.2. Chelating Agents and Cellular Viability
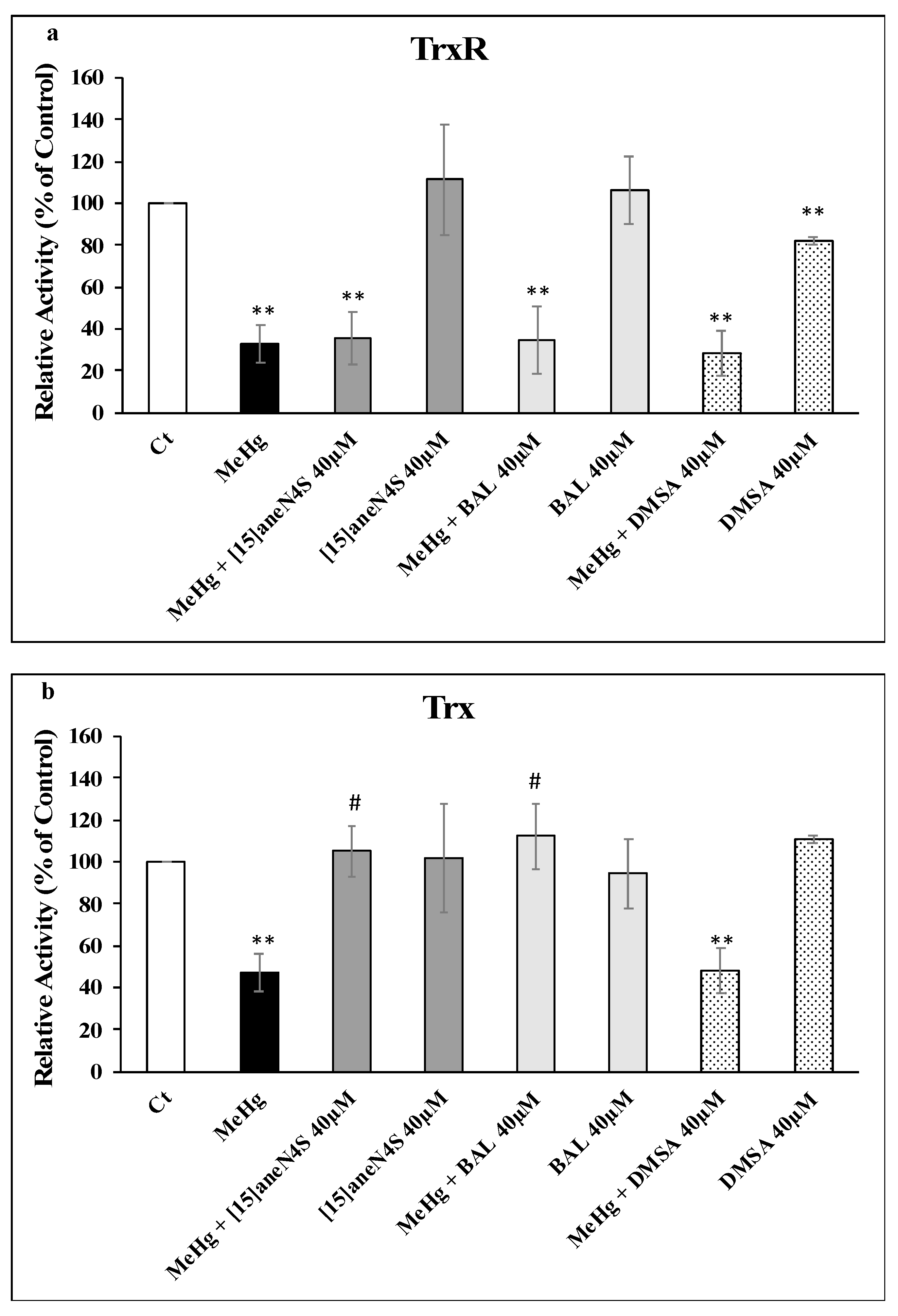
3.3. Thioredoxin Reductase and Thioredoxin Activity and Expression in SH-SY5Y Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Branco, V.; Caito, S.; Farina, M.; Teixeira da Rocha, J.; Aschner, M.; Carvalho, C. Biomarkers of mercury toxicity: Past, present, and future trends. J. Toxicol. Environ. Health Part B 2017, 20, 119–154. [Google Scholar] [CrossRef]
- Clarkson, T.W.; Magos, L. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 2006, 36, 609–662. [Google Scholar] [CrossRef]
- Antunes dos Santos, A.; Appel Hort, M.; Culbreth, M.; López-Granero, C.; Farina, M.; Rocha, J.B.T.; Aschner, M. Methylmercury and brain development: A review of recent literature. J. Trace Elem. Med. Biol. 2016, 38, 99–107. [Google Scholar] [CrossRef]
- Carvalho, C.M.L.; Matos, A.I.N.M.; Mateus, M.L.; Santos, A.P.M.; Batoréu, M.C.C. High-Fish Consumption and Risk Prevention: Assessment of Exposure to Methylmercury in Portugal. J. Toxicol. Environ. Health Part A 2008, 71, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Caetano, T.; Branco, V.; Cavaco, A.; Carvalho, C. Risk assessment of methylmercury in pregnant women and newborns in the island of Madeira (Portugal) using exposure biomarkers and food-frequency questionnaires. J. Toxicol. Environ. Health Part A 2019, 82, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Aschner, M.; Rocha, J.B.T. Oxidative stress in MeHg-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2011, 256, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Lee, E.; Ni, M.; Jiang, H.; Milatovic, D.; Rongzhu, L.; Farina, M.; Rocha, J.B.T.; Aschner, M. Methylmercury-induced alterations in astrocyte functions are attenuated by ebselen. Neurotoxicology 2011, 32, 291–299. [Google Scholar] [CrossRef]
- Rooney, J.P.K. The role of thiols, dithiols, nutritional factors and interacting ligands in the toxicology of mercury. Toxicology 2007, 234, 145–156. [Google Scholar] [CrossRef]
- Roos, D.H.; Puntel, R.L.; Lugokenski, T.H.; Ineu, R.P.; Bohrer, D.; Burger, M.E.; Franco, J.L.; Farina, M.; Aschner, M.; Rocha, J.B.T.; et al. Complex methylmercury-cysteine alters mercury accumulation in different tissues of mice. Basic Clin. Pharmacol. Toxicol. 2010, 107, 789–792. [Google Scholar] [CrossRef]
- Carvalho, C.M.L.; Chew, E.H.; Hashemy, S.I.; Lu, J.; Holmgren, A. Inhibition of the human thioredoxin system: A molecular mechanism of mercury toxicity. J. Biol. Chem. 2008, 283, 11913–11923. [Google Scholar] [CrossRef]
- Branco, V.; Carvalho, C. The thioredoxin system as a target for mercury compounds. Biochim. Biophys. Acta BBA Gen. Subj. 2019, 1863, 129255. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989, 264, 13963–13966. [Google Scholar] [PubMed]
- Lillig, C.H.; Holmgren, A. Thioredoxin and Related Molecules–From Biology to Health and Disease. Antioxid. Redox Signal. 2006, 9, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.L.; Lu, J.; Zhang, X.; Arnér, E.S.J.; Holmgren, A. Effects of selenite and chelating agents on mammalian thioredoxin reductase inhibited by mercury: Implications for treatment of mercury poisoning. FASEB J. 2011, 25, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Branco, V.; Godinho-Santos, A.; Gonçalves, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mitochondrial thioredoxin reductase inhibition, selenium status, and Nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic. Biol. Med. 2014, 73, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Branco, V.; Canário, J.; Holmgren, A.; Carvalho, C. Inhibition of the thioredoxin system in the brain and liver of zebra-seabreams exposed to waterborne methylmercury. Toxicol. Appl. Pharmacol. 2011, 251, 95–103. [Google Scholar] [CrossRef]
- Branco, V.; Canário, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mercury and selenium interaction in vivo: Effects on thioredoxin reductase and glutathione peroxidase. Free Radic. Biol. Med. 2012, 52, 781–793. [Google Scholar] [CrossRef]
- Branco, V.; Coppo, L.; Solá, S.; Lu, J.; Rodrigues, C.M.P.; Holmgren, A.; Carvalho, C. Impaired cross-talk between the thioredoxin and glutathione systems is related to ASK-1 mediated apoptosis in neuronal cells exposed to mercury. Redox Biol. 2017, 13, 278–287. [Google Scholar] [CrossRef]
- Nunes, E.; Cavaco, A.; Carvalho, C. Exposure assessment of pregnant Portuguese women to methylmercury through the ingestion of fish: Cross-sectional survey and biomarker validation. J. Toxicol. Environ. Health A 2014, 77, 133–142. [Google Scholar] [CrossRef]
- Bjørklund, G.; Mutter, J.; Aaseth, J. Metal chelators and neurotoxicity: Lead, mercury, and arsenic. Arch. Toxicol. 2017, 91, 3787–3797. [Google Scholar] [CrossRef]
- Crea, F.; Stefano, C.; Foti, C.; Milea, D.; Sammartano, S. Chelating Agents for the Sequestration of Mercury(II) and Monomethyl Mercury(II). Curr. Med. Chem. 2014, 21, 3819–3836. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Skaug, M.A.; Andersen, O.; Aaseth, J. Chelation therapy in intoxications with mercury, lead and copper. J. Trace Elem. Med. Biol. 2015, 31, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Delgado, R.; Félix, V.; Lima, L.M.P.; Price, D.W. Metal complexes of cyclen and cyclam derivatives useful for medical applications: A discussion based on thermodynamic stability constants and structural data. Dalton Trans. 2007, 2734–2745. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.S.; Cabral, M.F.; Costa, J.; Castro, M.; Delgado, R.; Drew, M.G.B.; Félix, V. Two macrocyclic pentaaza compounds containing pyridine evaluated as novel chelating agents in copper(II) and nickel(II) overload. J. Inorg. Biochem. 2011, 105, 410–419. [Google Scholar] [CrossRef]
- Mewis, R.E.; Archibald, S.J. Biomedical applications of macrocyclic ligand complexes. Coord. Chem. Rev. 2010, 254, 1686–1712. [Google Scholar] [CrossRef]
- Torres, N.; Gonçalves, S.; Fernandes, A.S.; Machado, J.F.; De Brito, M.J.V.; Oliveira, N.G.; Castro, M.; Costa, J.; Cabral, M.F. [15]aneN4S: Synthesis, thermodynamic studies and potential applications in chelation therapy. Molecules 2014, 19, 550–567. [Google Scholar] [CrossRef]
- Arnér, E.S.; Holmgren, A. Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol. 2001, 24, 7–14. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Zhong, L.; Arnér, E.S.; Holmgren, A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA 2000, 97, 5854–5859. [Google Scholar] [CrossRef]
- Flora, S.J.S.; Pachauri, V. Chelation in metal intoxication. Int. J. Environ. Res. Public Health 2010, 7, 2745–2788. [Google Scholar] [CrossRef]
- Bjørklund, G.; Crisponi, G.; Nurchi, V.M.; Cappai, R.; Djordjevic, A.B.; Aaseth, J. A Review on Coordination Properties of Thiol-Containing Chelating Agents Towards Mercury, Cadmium, and Lead. Molecules 2019, 24, 3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nobre, P.; Cabral, M.d.F.; Costa, J.; Castro-Caldas, M.; Carvalho, C.; Branco, V. In Vitro Assessment of the Efficacy of a Macrocyclic Chelator in Reversing Methylmercury Toxicity. Int. J. Environ. Res. Public Health 2019, 16, 4817. https://doi.org/10.3390/ijerph16234817
Nobre P, Cabral MdF, Costa J, Castro-Caldas M, Carvalho C, Branco V. In Vitro Assessment of the Efficacy of a Macrocyclic Chelator in Reversing Methylmercury Toxicity. International Journal of Environmental Research and Public Health. 2019; 16(23):4817. https://doi.org/10.3390/ijerph16234817
Chicago/Turabian StyleNobre, Paula, Maria de Fátima Cabral, Judite Costa, Margarida Castro-Caldas, Cristina Carvalho, and Vasco Branco. 2019. "In Vitro Assessment of the Efficacy of a Macrocyclic Chelator in Reversing Methylmercury Toxicity" International Journal of Environmental Research and Public Health 16, no. 23: 4817. https://doi.org/10.3390/ijerph16234817
APA StyleNobre, P., Cabral, M. d. F., Costa, J., Castro-Caldas, M., Carvalho, C., & Branco, V. (2019). In Vitro Assessment of the Efficacy of a Macrocyclic Chelator in Reversing Methylmercury Toxicity. International Journal of Environmental Research and Public Health, 16(23), 4817. https://doi.org/10.3390/ijerph16234817