1. Introduction
One of the foremost public health problems affecting the world today is smoking, which represents a preventable cause of premature death [
1]. Smoking has a crucial implication in causing many common diseases such as cancer, chronic obstructive pulmonary diseases, and periodontitis [
2,
3,
4]. The impact of smoking tobacco on periodontal health is not a novel concept; however, several studies have shown the significant adverse effect of smoking on the microbiome and cytokines expression of the buccal mucosa [
5]. Nevertheless, tobacco in addition to other environmental factors may affect the equilibrium of the oral microbiome by inducing a possible alteration in functional pathways and allowing oral pathogens to grow, which ultimately leads to several diseases [
6,
7,
8]. Furthermore, different oral pathogens harm the development and function of innate and adaptive immunity of the host [
9]. The ease of saliva collection indicates that saliva microbiome studies have a vital implication in disease diagnosis or prognosis [
10]. Jordan is one of the many countries dealing with the smoking epidemic. A cross-sectional survey was conducted among 11 to 18-year-old school students, including boys and girls, from an important governorate in Jordan. The study surprisingly showed a high rate of smoking and particularly dual tobacco consumption, cigarettes, and waterpipe [
11]. The studies emphasized that there is an increasing smoking rate among the Jordanian population, especially among the youth, which indicates awareness insufficiency of the destructive effects of smoking and the necessity for prevention programs to handle such knowledge deficiency. Therefore, this study is to investigate the impact of smoking on oral cavity microbiome components among adult Jordanian smokers and the comparison between smoking and non-smoking subjects by using high-throughput 16S rRNA gene sequencing via next-generation sequencing technology (NGS). The outcome of the current study is expected to help identify the interactions between smoking and the salivary microbiome.
The oral microbiome is an essential player that induces a dynamic equilibrium with the immune-inflammatory response of the host [
12]. The human oral cavity is one of the entries of the respiratory tract, and the main entry point for several microorganisms, primarily airborne pathogens, and those transferred through saliva. The salivary microbiome possesses its characteristic microorganisms and interacts with other microbiomes in the human body, especially that of the intestinal tract [
13]. There is excellent genera diversity in the human salivary microbiome; therefore, it is essential to understand the role of these known and unknown genera in the oral cavity and how they interact with the microbiomes of other systems in the human body [
13].
The precision in determining the oral microbiome is not easily attainable because the oral cavity is an open system exposed continuously to bacteria present in food and water in addition to bacteria contracted through social contact. It is challenging to determine whether existing colonizations are a long-term diversity or not. A diversity that makes this community able to provide an appropriate response to each environmental stress or factors such as smoking, diet, oral hygiene, and drug consumption, e.g., antibiotics. Tobacco smoking generates carcinogens that contain distinct nitrosamines and free radicals capable of inhibiting antioxidant enzymes. In turn, an inhibited antioxidant enzyme makes the oral epithelial cells unprotected against the damaging effects of thiocyanate ions and hydroxyl free radicals. Thiocyanate ions and free radicals could react with DNA, adversely, and therefore, open the gateway to the progression of oral cancer [
14]. By inhibiting granulocyte function, smoking impairs host defenses and affects the immune system [
15].
Furthermore, subsequent nicotine metabolites trigger vasoconstriction and prejudice the role of polymorphonuclear cells and macrophages as well as decrease the number of lymphocytes, which may adversely affect the production of B-cells and antibodies [
16]. Furthermore, smoking contributes to the increase in the number of neutrophils in peripheral blood [
17]. The changes that occur as a result of activating inflammatory cells, which leads to the release of free radicals, were found to influence a move to malignancy by lipids’ peroxidation or DNA damage. The phyla
Firmicutes,
Bacteroidetes,
Proteobacteria,
Actinobacteria,
Spirochaetes, and
Fusobacteria dominate the oral cavity, accounting for more than 95% of the species [
18]. Various health-associated bacteria have been known to be antagonistic to oral pathogens;
Streptococcus salivarius strain K12, for example, produces a bacteriocin that prevents the growth of Gram-negative species linked to periodontitis [
19].
2. Materials and Methods
2.1. Study Subjects
One hundred (n = 100) human subjects participated in this study; 57 were males and 43 were females. According to the smoking status, 51 were non-smokers and 49 were smokers. The inclusion criteria required that all human subjects were antibiotic-free for the last three months preceding the study by ensuring that no one has consumed antibiotics in that period. Inclusion criteria for smokers required that all smoker subjects smoked at least one cigarette per day. The exclusion criteria, on the other hand, required the rejection of human subjects who had a history of any chronic oral diseases.
Additionally, saliva collection from all subjects was taken half an hour before, or an hour after eating. Signed informed consent and answered questions were obtained from all participants in this study according to the declaration of Helsinki. The Council of Scientific Research at the German Jordanian University has approved the proposal of the study based on decision #31/3/2016 as stated in letter #389/6/4/10.
2.2. Sample Collection, Processing, and Storage
All human subjects had to spit their unstimulated saliva into the OMNIgene•ORAL OM-501™ funnel, which is commercially available by DNA Genotek, ON, Canada. Subjects kept on spitting until the amount of spat liquid, excluding bubbles, reached the filled line mark indicated on the wall of the collecting tube. All human subjects were required to hold the collecting tubes upright with one hand and close the funnel lid with the other hand. A liquid DNA stabilizer, placed in the tube cover, was automatically released at this stage into the tube after replacing the funnel with the tube cap to firmly close the collecting tube. The DNA stabilizer stabilizes the microbial DNA in saliva for up to one year at room temperature. The DNA stabilizer was then mixed with the collected liquid sample for 10 s. The samples were shipped at room temperature to DNA Genotek GenoFIND Services, Norcross, GA, USA, for complete processing.
2.3. DNA Extraction and Quality Controls
A 250 µL aliquot of each sample was extracted using MO BIO’s PowerMag™ microbial DNA isolation kit (27200-4) (MO BIO Laboratories Inc., Carlsbad, CA, USA) optimized on the KingFisher automated extraction platform. A proprietary bead-beating step with glass beads and a plate shaker was used to maximize recovery of DNA from low-abundance and challenging to lyse organisms. The concentration of extracted DNA was determined by Qubit measurement, and an estimate of sample purity was determined with spectrophotometry by measuring the A260/A280 absorbance ratio. Quality control checks are tabulated in
Appendix A data (
Table A1).
2.4. DNA Sequencing
Illumina sequencing adapters and dual-index barcodes (Nextera XT indices) were added to the amplicon target via polymerase chain reaction (PCR) amplification. Samples were run on Bioanalyzer, spot-checking for amplicon size. The 16S sequencing (2 × 300 bp PE V3-V4) was performed on Illumina’s MiSeq platform (Illumina Inc., San Diego, CA, USA). Paired-end reads from each sample were merged, screened for length, and filtered for quality using DNA Genotek’s proprietary 16S pre-processing workflow. The sequence data were submitted to NCBI BioProject under accession number PRJNA579773.
2.5. Taxonomic Classification
High-quality sequences were aligned to the curated reference database at 97% similarity using the NINJA-OPS algorithm, version 1.5.1 [
20]. At 97% sequence identity, each operational taxonomic unit (OTU) represents a genetically unique group of biological organisms. These OTUs were then assigned a curated taxonomic label based on the SILVA taxonomic database, version 123 [
21]. The relative abundance of all taxa at the phylum and genus levels were plotted to visualize broad taxonomic differences between individual samples and between sample groups. Genera found at <1% mean abundance across samples were grouped as “other” for visualization purposes. MicrobiomeAnalyst, a web-based data analysis tool, was chosen to perform Univariate statistical analysis for features at the phyla and genera levels; features were considered significant based on their adjusted cut-off ≤ 0.05. This web-based tool has been reported and is currently hosted by the Xia lab at McGill University, QC, Canada [
22].
2.6. Rarefaction
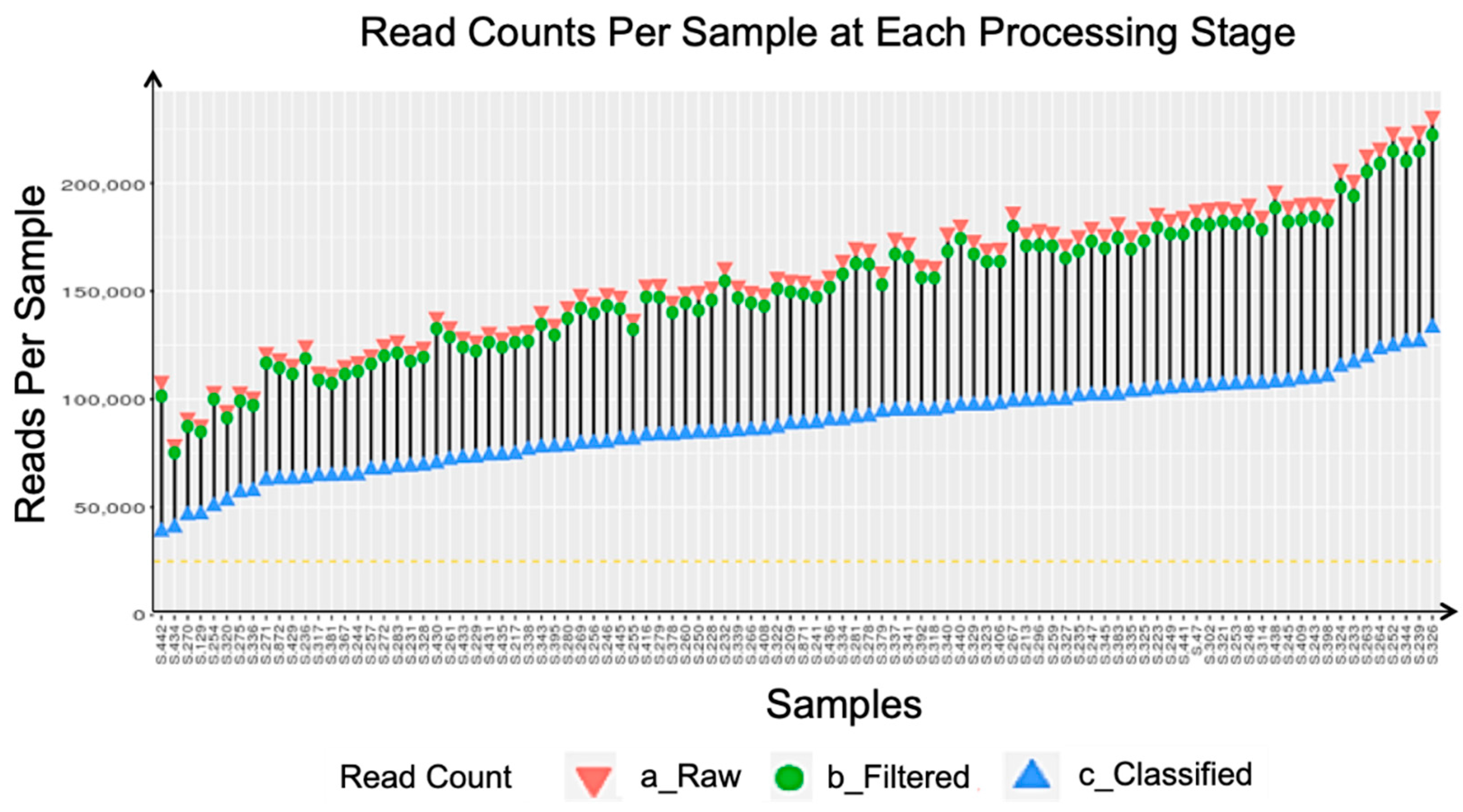
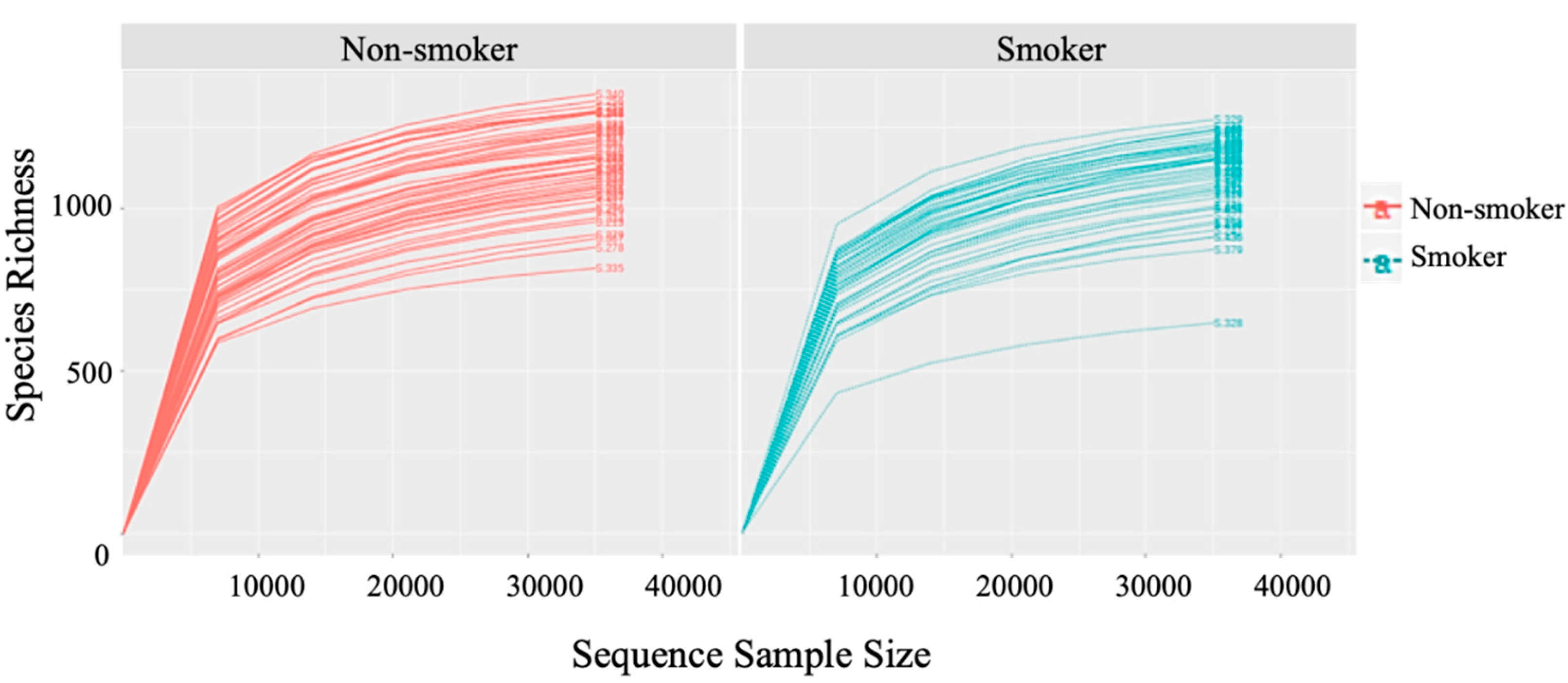
All samples were rarefied after taxonomic classification. The cutoff for rarefaction was set at 25,000 classified sequences per sample. However, no sample had less than 25,000 classified sequences, thus, all samples were included in the downstream analysis (see
Figure A1 and
Figure A2).
2.7. Library Preparation and Sequence Amplification
Library preparation was performed with a customized dual index version of Illumina’s Nextera XT protocol. The V3-V4 region of the 16S ribosomal subunit was amplified with custom polymerase chain reaction (PCR) primers and sequenced on an Illumina MiSeq.
2.8. Data Pre-Processing
Trimmomatic was used to remove sequencing adaptors, and low-quality reads [
23]. The FLASH algorithm was used to read merging and automated rejection of low-quality sequences [
24]. Quality screening for length and ambiguous bases was performed with proprietary scripts [
23].
2.9. Data Analysis
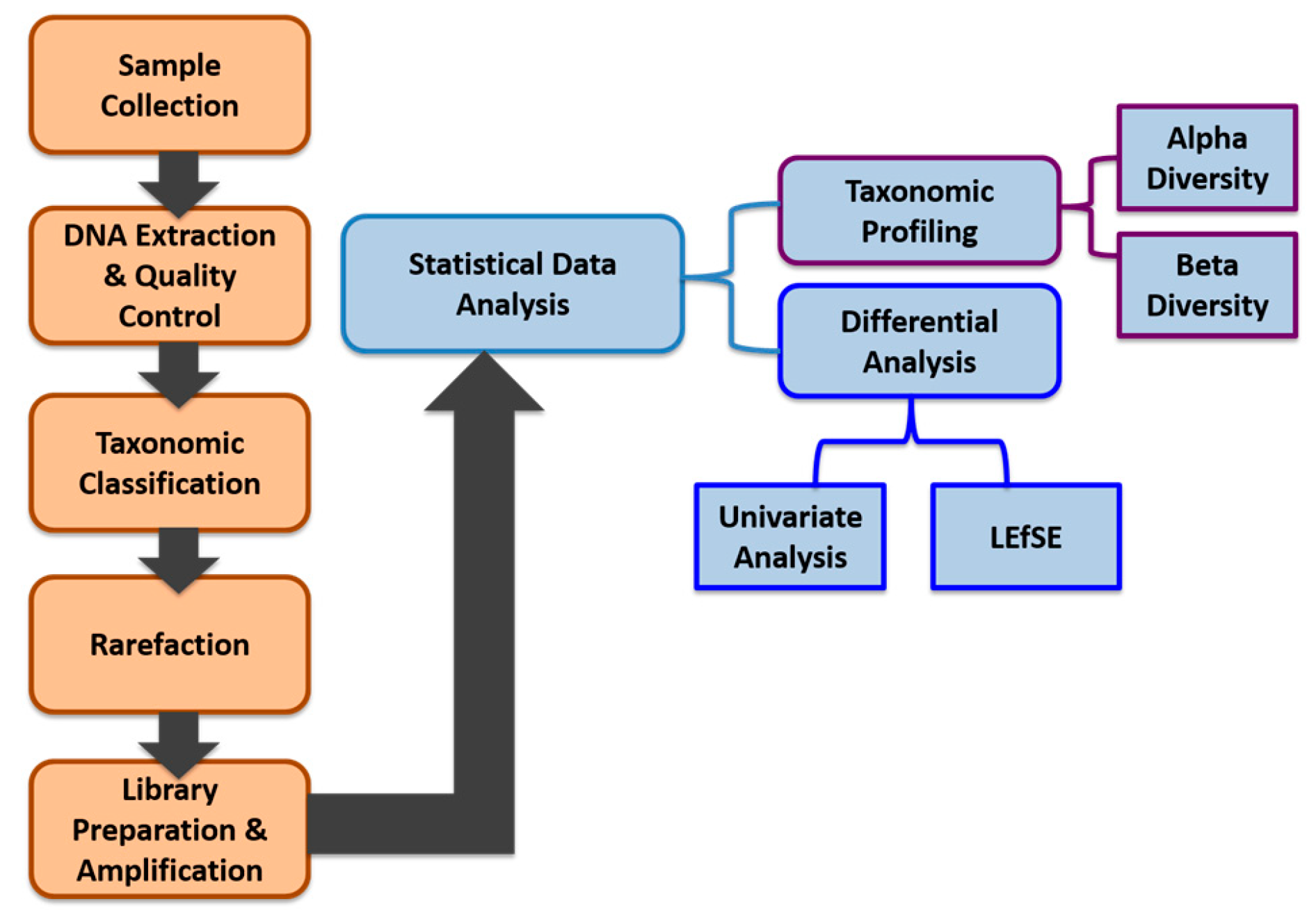
We applied a comprehensive bioinformatics analysis approach integrating both robust exploratory data analysis and visualization methods focusing on taxonomic profiling, combined with standard statistical differential analysis approaches such as univariate analysis methods to identify statistically significant features in terms of their abundance between different smokers and non-smokers. We also applied the linear discriminant analysis (LDA) effect size (LEfSe) method to support high-dimensional class comparisons. Our data generation, data preprocessing, and data analysis workflow is shown in
Figure 1.
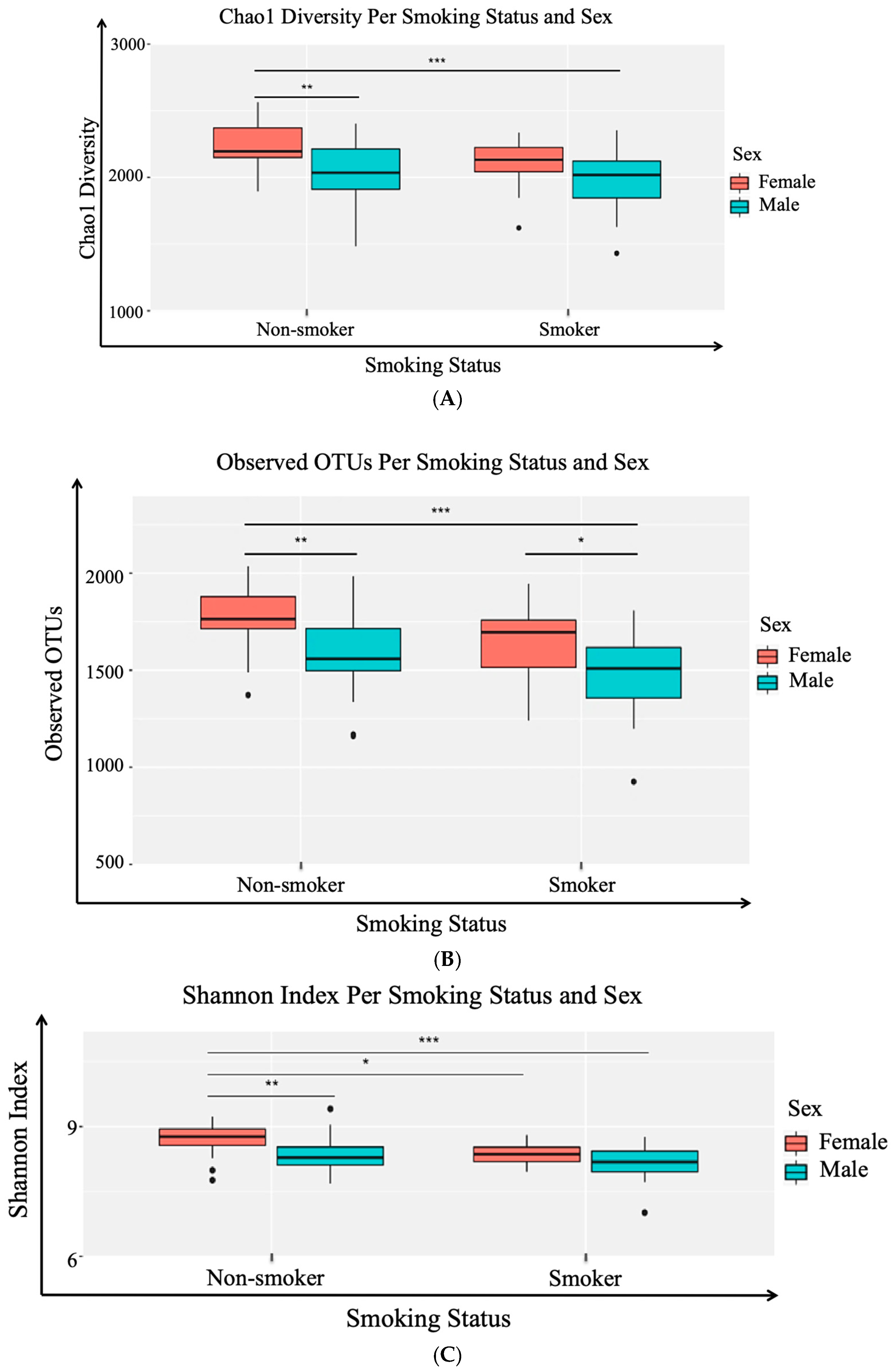
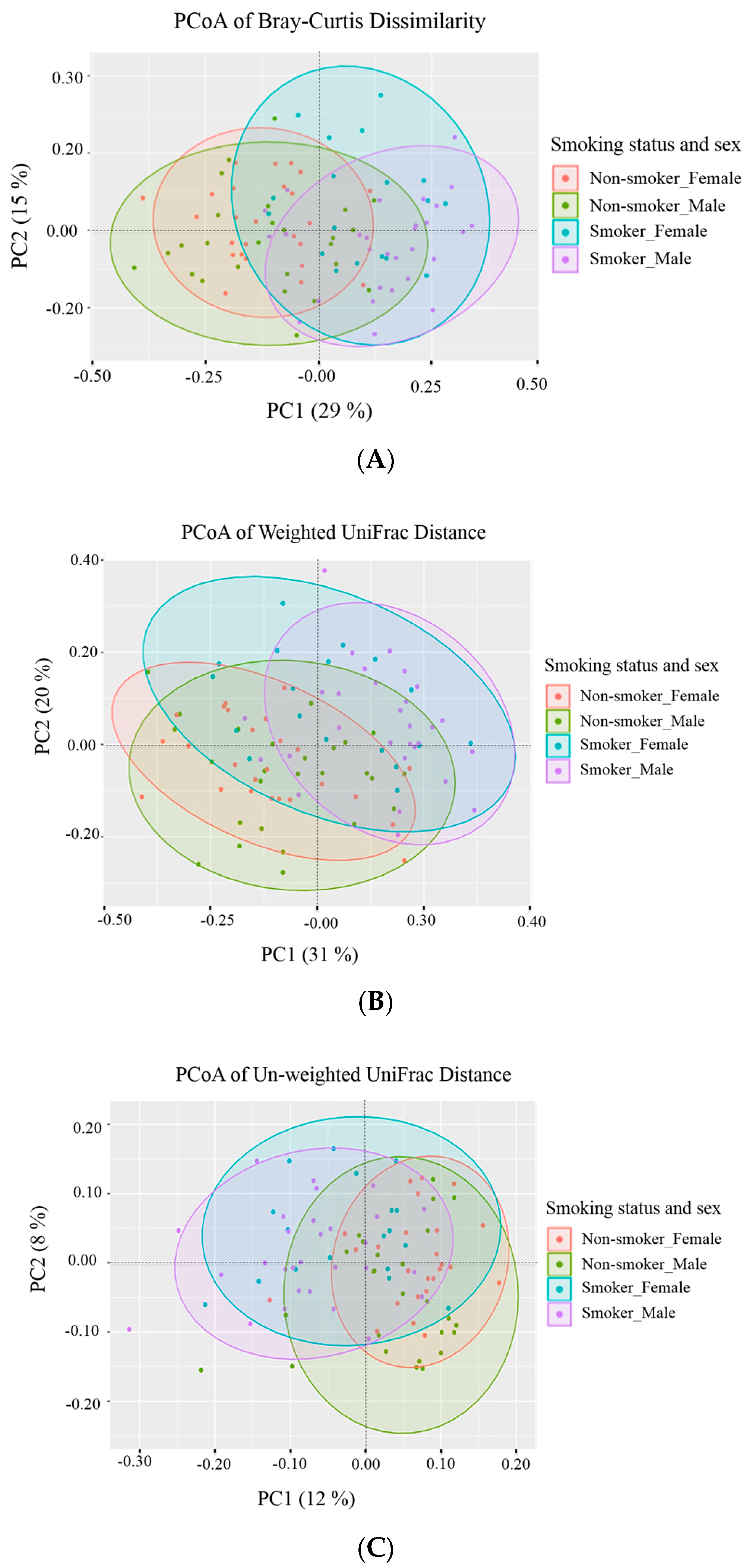
2.9.1. Alpha (α) Diversity and Beta (β) Diversity
Taxonomic profiling: exploratory data analysis and visualization consisted of two main methods: (a) alpha diversity analysis for assessing diversity within a bacterial community or sample and (b) beta diversity analysis for determining the differences between microbial communities (i.e., between samples). Three different alpha diversity metrics (Shannon Index, Observed OTUs, Chao1 diversity) were calculated on rarefied OTU tables using the
alpha_rarefaction.py workflow in QIIME 1.9.1 [
25] and the results were determined by using Analysis of Variance for each alpha diversity metric. Tukey’s honestly significant difference (HSD) was applied to the AOV for analysis of variance (ANOVA), it is an R function to determine group-to-group comparisons. The R version 3.3.2 (
R Core Team,
2015) was used to perform the statistical analyses of alpha and beta diversity. Additionally, three beta metrics were used (Bray-Curtis, Weighted UniFrac, and Unweighted UniFrac) on the rarefied OTU table using the
beta_diversity.py workflow in QIIME 1.9.1. Bray-Curtis dissimilarity was calculated on a species-level summarization of the rarefied OTU table [
26]. Principal Coordinates Analysis (PCoA) was applied to each beta diversity distance matrix, using the
dudi.pco function from the R
made4 package (version 1.48.0). The first two principal coordinates explaining the majority of the difference in data were plotted using R’s
ggplot2 package, version 2.2.1), with the indicated percentage of variance by each axis explained.
2.9.2. Univariate Analysis
Two standard univariate tests, implemented in MicrobiomeAnalyst [
22] from the Xia lab at McGill University in Canada, were applied to test for statistically significant abundant taxa between smokers and non-smokers. The tests were: (a) non-parametric Mann-Whitney test and (b) parametric t-test/ANOVA. Our differential analysis helps in identifying biologically or biochemically meaningful relationships or associations between taxa or features. The analyses were conducted at phylum and genus levels.
2.9.3. LDA Effect Size (LEfSe)
This method is specifically designed for biomarker discovery and explanation in high-dimensional metagenomic data [
27]. It incorporates statistical significance with biological consistency (effect size) estimation. It performs a non-parametric factorial Kruskal-Wallis (KW) sum-rank test to identify features with significant differential abundance with regard to experimental factor or class of interest, followed by Linear Discriminant Analysis (LDA) to calculate the effect size of each differentially abundant features. The result consists of all the features, the logarithmic value of the maximum mean among all the groups or classes, and if the features are differentially significant, the group with the highest mean and the logarithmic LDA score (Effect Size). Features are considered to be significant based on their adjusted
p-values (i.e., false discovery rate (FDR) values), applying an adjusted
p-value cutoff = 0.05.
4. Discussion
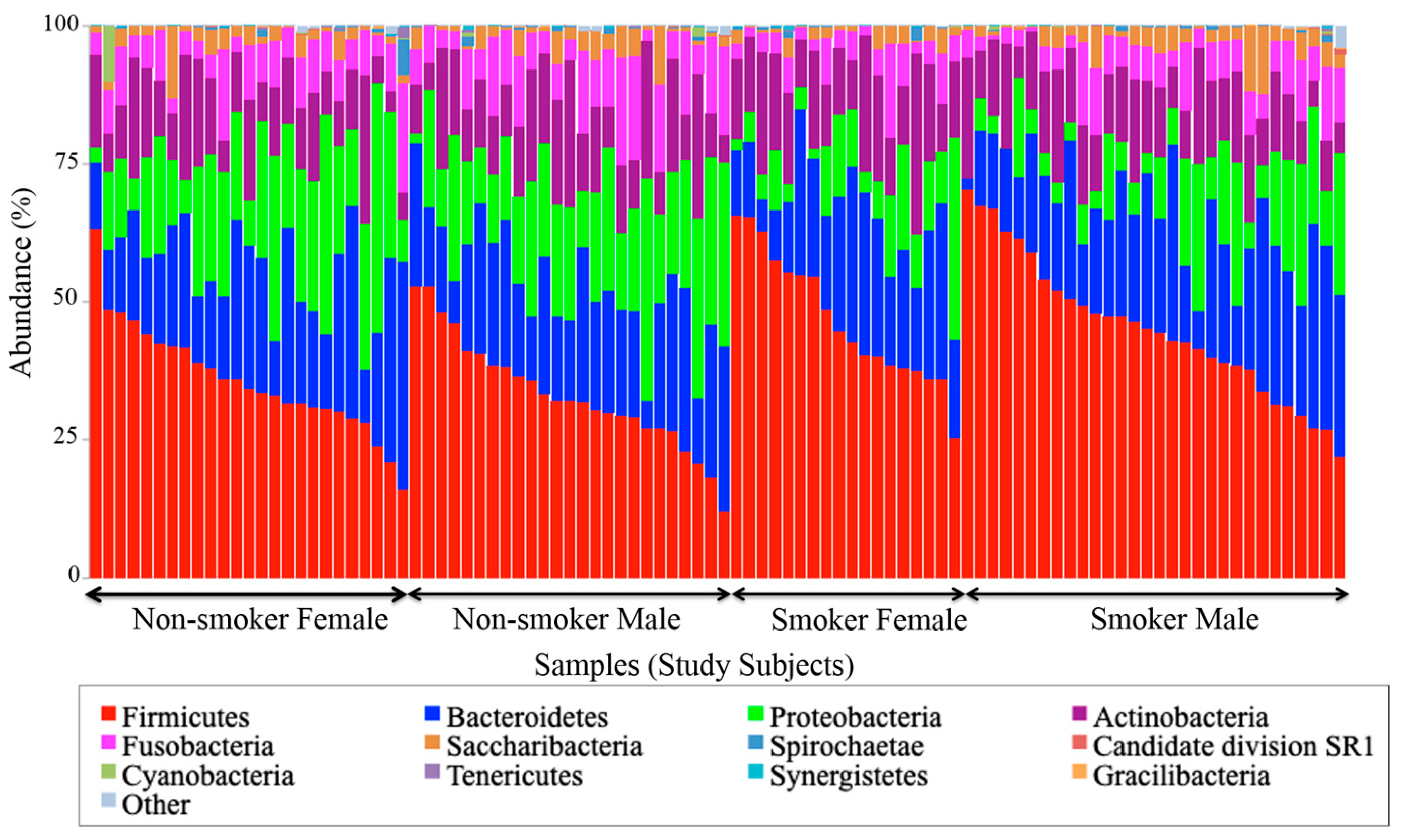
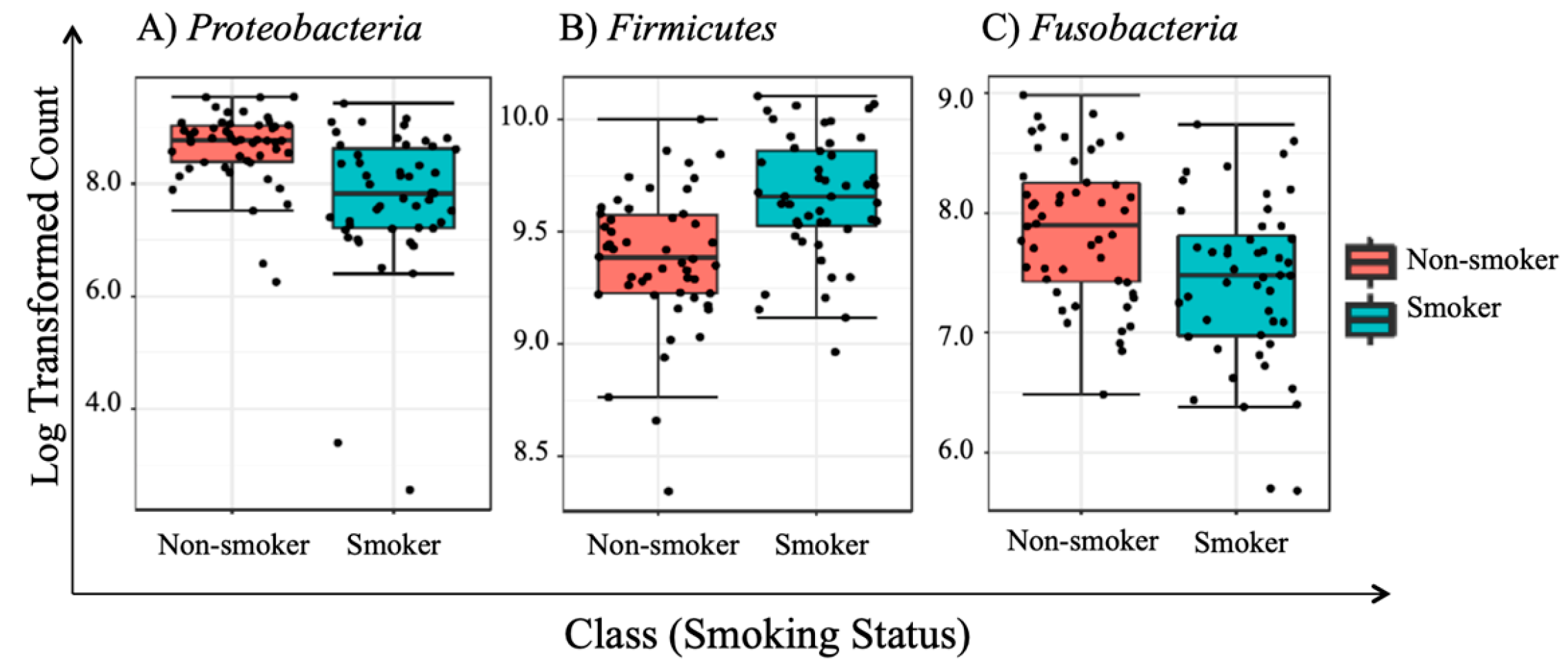
To the best of our knowledge, this paper represents a first of its kind report in Jordan, documenting statistically significant changes in the salivary human microbiome composition between smoker and non-smoker human subjects. Our methods relied on high throughput next-generation sequencing of the 16S rRNA marker gene determined in unstimulated salivary samples. Based on the outcomes of previous studies addressing the adverse effects of smoking on health in general, it was anticipated that pathogenic bacteria might be present at the expense of the commensal flora in smokers. This study showed that alpha and beta diversity displayed intra and inter-individual variations. However, the profile clustering direction for each study group (male smokers, female smokers, male non-smokers, and female non-smoker) was apparent with interesting overlapping Venn diagrams for male and female non-smokers versus male and female smokers. This implies a significant response to smoking regardless of gender, even with slight significant statistical variation between males and females in general. Firmicutes, Proteobacteria, and Bacteroidetes were found to have the highest relative abundance percentage of the community at phylum level in all samples. However, smoking had affected the Firmicutes, Proteobacteria, and Fusobacteria, as Firmicutes was statistically elevated in smokers at the expense of Proteobacteria and Fusobacteria in non-smokers. This implies that smoking has a critical impact on the homeostasis of human salivary microbiome. The biological meaning of these findings was not evident until we performed the analysis at genera level.
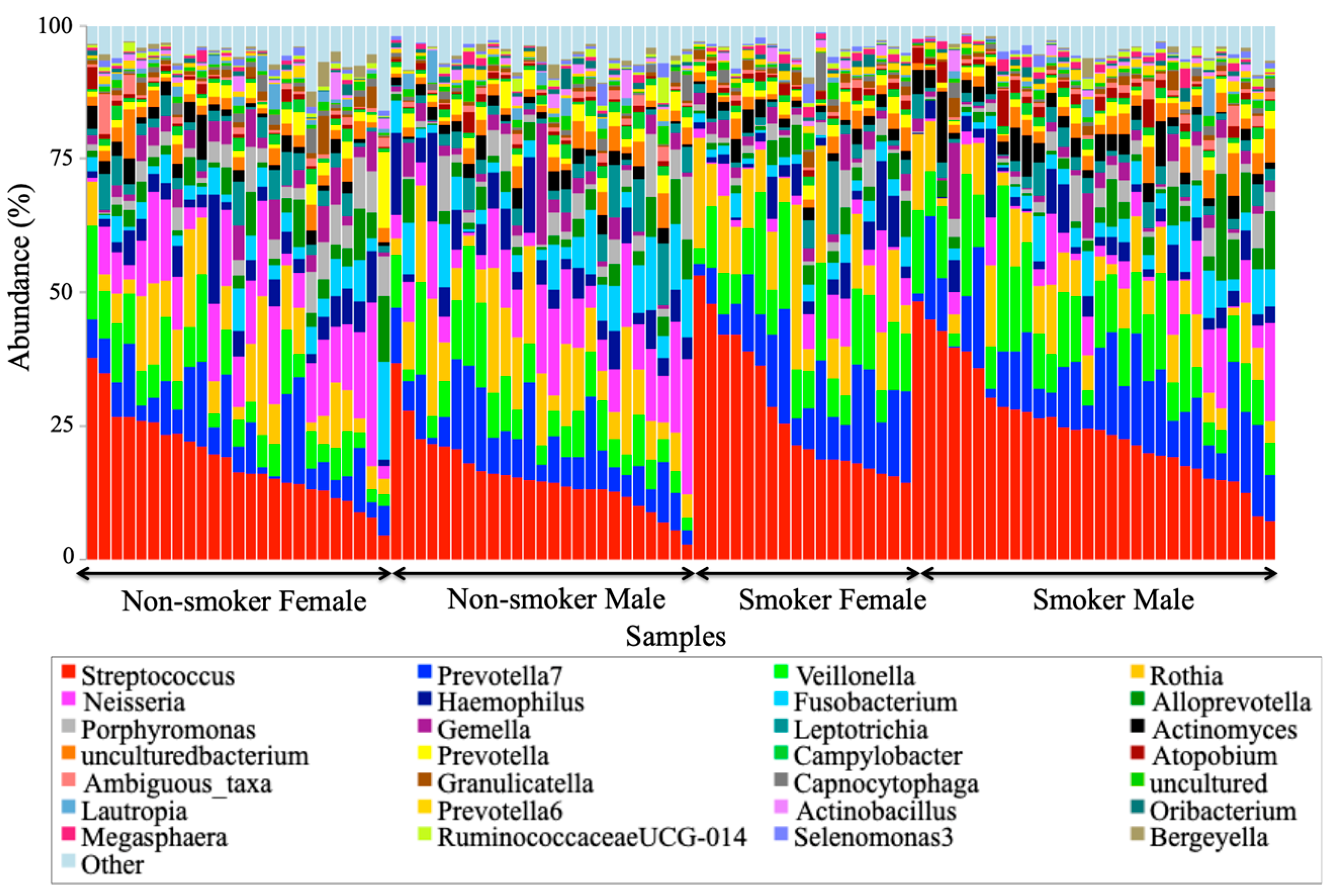
At the genus level,
Streptococcus,
Prevotella,
Vellionella,
Rothia,
Neisseria, and
Haemophilus predominated the salivary microbiota of all examined samples.
Streptococcus,
Prevotella, and
Veillonella were the most significantly predominant genera among smokers at the expense of
Neisseria that are healthy flora in the human oral cavity, which has been significantly decreased among smokers. The increased levels of
Streptococcus and
Veillonella and the reduced level of
Neisseria were consistent with an extensive study of in a thorough survey of cigarette smoking and oral microbiome among American adults [
8]. The reduced level of anaerobic
Neisseria in this study is consistent with a human oral microbiota study [
29], which might be related to the effect of oxygen deprivation in the oral cavity caused by smoking. The predominance of the anaerobic
Veillonella and the facultative anaerobic
Streptococcus may explain their success in tolerating the lack of oxygen in the smoking microenvironment. Elevated
Prevotella was correlated to oral malodor (halitosis) [
30], which can be caused by smoking in this study, which is consistent with a clinical review by Porter and Scully [
31].
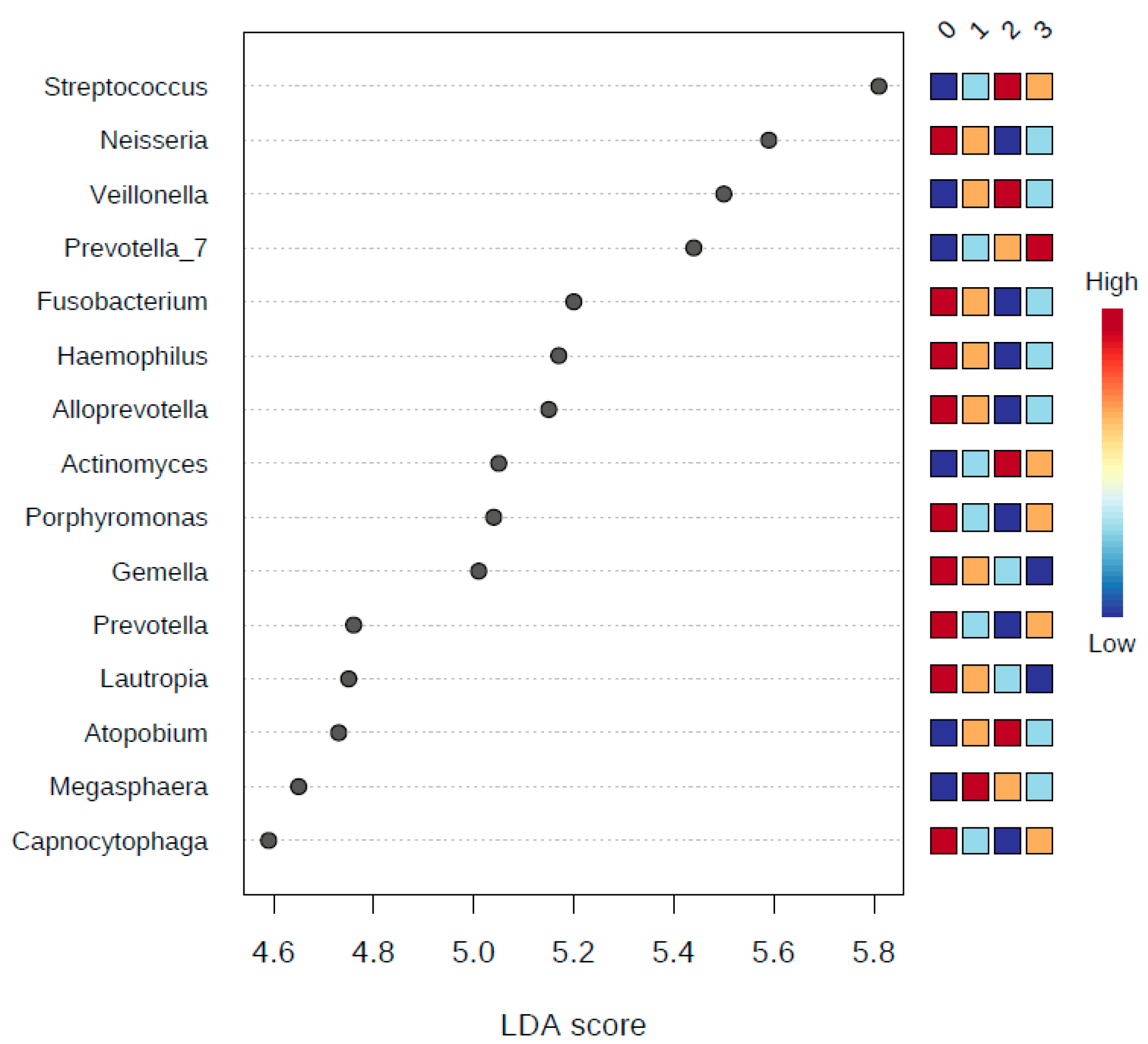
Since statistically significant taxa do not always convey the biological messages, we want to arrive in to make important discoveries later on. We performed additional statistical tests to build confidence in the prioritized taxa and make sure that these taxa are able to explain the differences between the studies classes of smokers and non-smokers. This, in addition to a subsequent related classification based on the number of years of smoking, the number of cigarettes smoked, and whether the human subjects brush or did not brush their teeth, in terms of teeth brushing the phylum
Synergistetes were identified as a statistically significant phylum distinguishing the human subjects that brush and those that do not brush their teeth.
Synergistetes has been reported in both periodontal health and disease [
32]; thus, a further investigation at the species level of
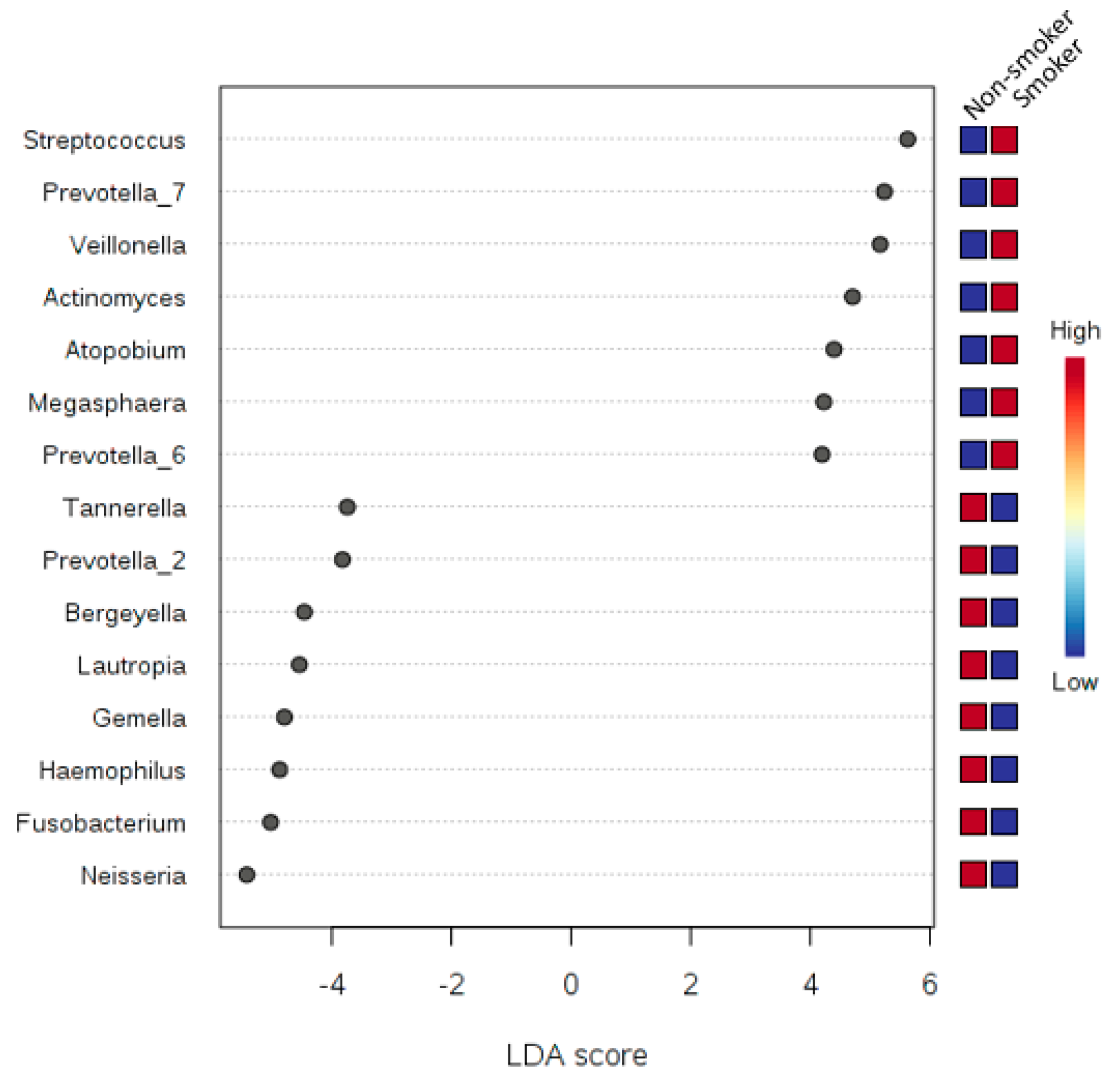
Synergistetes is needed. Our tests relied on LEfSe, which combines standard tests for statistical significance with additional tests encoding biological consistency and effect relevance. Based upon our LEfSe results analysis in
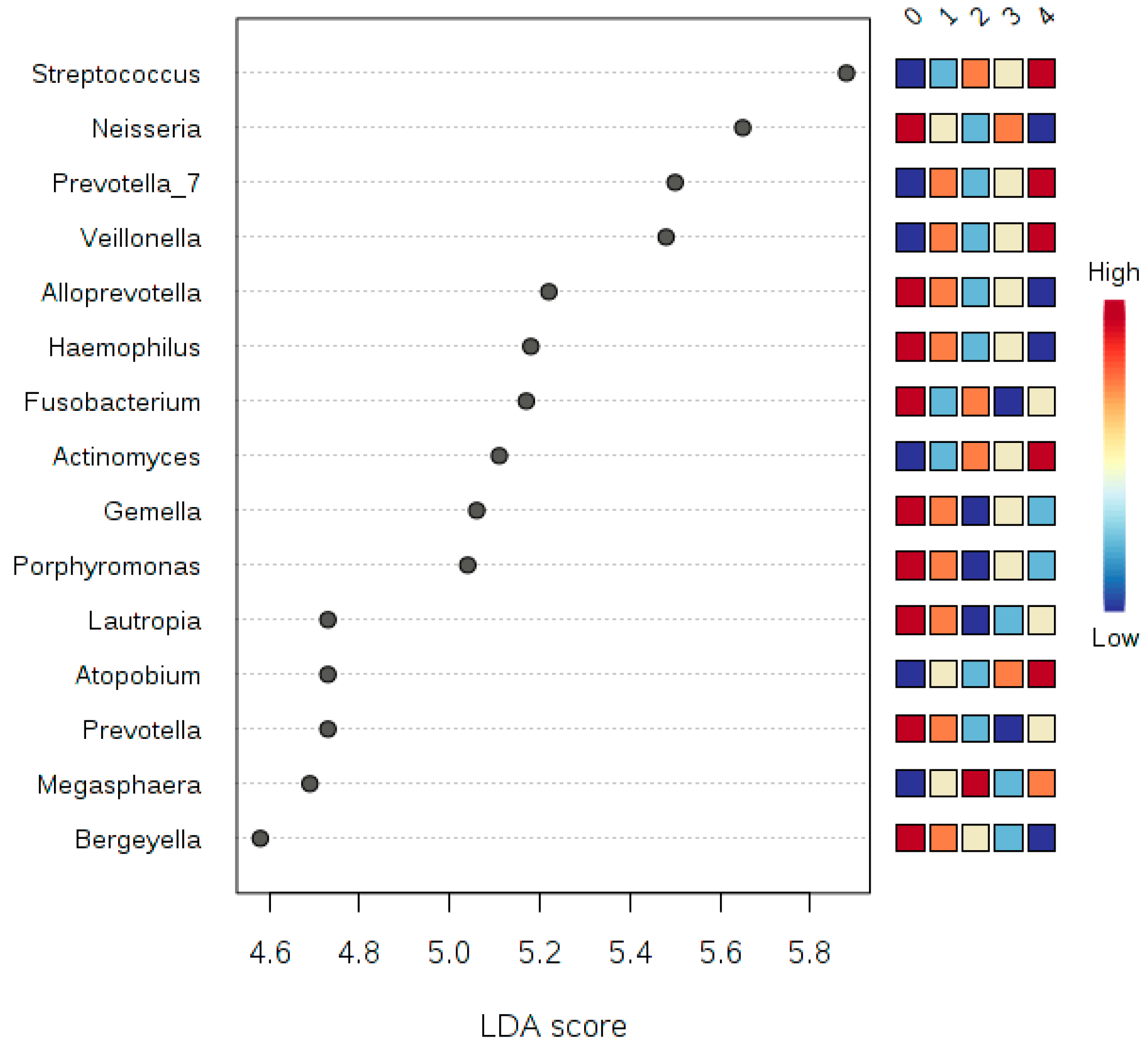
Figure 7,
Figure 8 and
Figure 9, we can see that there is a microbial signature distinguishing smokers from non-smokers, which is consistent with our univariate analysis except that the abundance of
candidate division SR1 was statistically significant (
Table 5). The
candidate division SR1 usually described in the literature as unknown or unaffiliated [
33]. We did not see such a clear signature distinguishing the different classes of human subjects resulting from the binned numbers of years of smoking and the binned numbers of the number of cigarettes smoked.
We can suggest that there is microbial signature at the genera level can be used to classify smokers and non-smokers by LEfSe based on the salivary abundance of the 15 genera including, but not limited to, Streptococcus, Prevotella, and Veillonella, which are all more abundance in smokers relative to non-smokers, and Neisseria, which is more abundant in non-smokers relative to smokers.
It is worthy to note that infections are believed to be a cause of carcinogenesis, alongside other known risk factors such as smoking tobacco and consuming alcohol. The case for role infections in carcinogenesis is increasingly solidified with evidence that the inflammation bacteria can secrete endotoxins, which in turn might induce DNA damage in mouth epithelial tissue [
34,
35]. A positive correlation between proinflammatory cytokine levels and commensal bacteria was observed in smokers, but that correlation was not present for non-smokers. A previous study suggested that smoking affects both the composition of the nascent biofilm and the host reaction to this colonization [
3]. The elevated abundance of
Streptococcus,
Prevotella, and
Veillonella in this study should be considered in future research to explore the feasibility of being a salivary diagnostic predictor in Jordanian smokers for oral squamous cell carcinomas. For example, a previous report concluded that some specific taxa have a significant correlation with epithelial precursor regions and oral cancer, taxa such as
Streptococcus spp.,
Veillonella,
Porphyromonas,
Fusobacterium,
Prevotella,
Actinomyces,
Clostridium,
Haemophilus, and
Enterobacteriaceae [
36].
Although we were successful in generating high-quality sequencing data that enabled the subsequent bioinformatics analysis to identify significant compositional differences in the salivary microbiome between smokers and non-smokers, this study has some limitations that we should disclose here. First, targeting only two hypervariable regions, V3-V4 on the 16S rRNA gene, might group closely related taxa into a single taxonomic unit. However, there is sufficient evidence in the biomedical literature indicating that the V3-V4 exploration is adequate to produce a reliable phylogenetic ranking at phyla and genus levels, but usually not at species level. Second, even though 16S rRNA remains the most efficient available approach to study microbial communities, it suffers mosaicism, intra-genomic heterogeneity, and lacks a universal threshold of what is known as sequence identity value [
37]. Third, we could not control for other confounders for example lifestyle, exact and complete oral health beyond yes or no teeth brushing, alcohol consumption, drug abuse, and chemical and physical properties of saliva. Last, it is challenging to determine whether the existing microbial colonization is a long-term one or not.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










