Identification of Potential Biomarkers and Related Transcription Factors in Peripheral Blood of Tuberculosis Patients



Abstract
:1. Introduction
2. Material and Methods
2.1. Microarray Data
2.2. Data Processing
2.3. GO and KEGG Pathway Enrichment Analysis
2.4. Protein–Protein Interaction (PPI) Network Construction and Module Analysis
2.5. TFs Regulatory Network of Hub Genes
3. Results
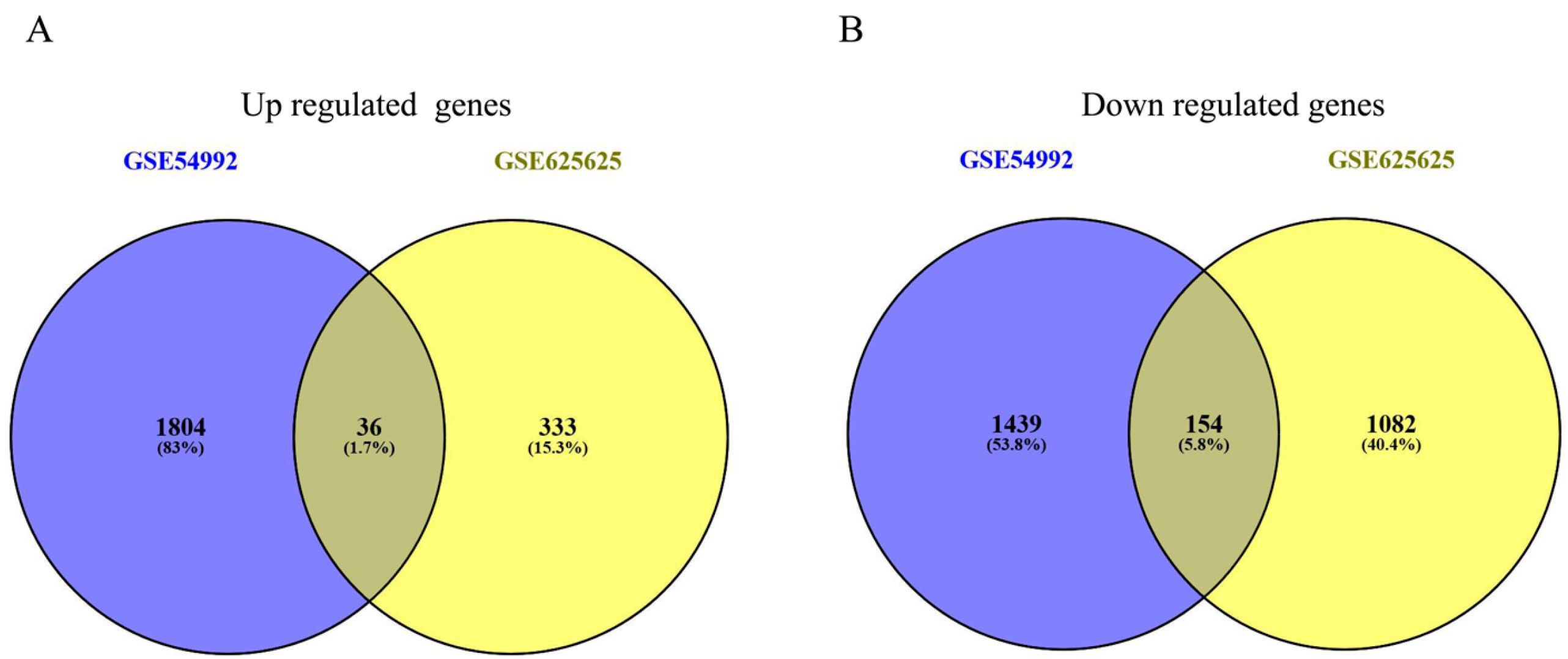
3.1. Identification of DEGs in PBMC from TB Patients
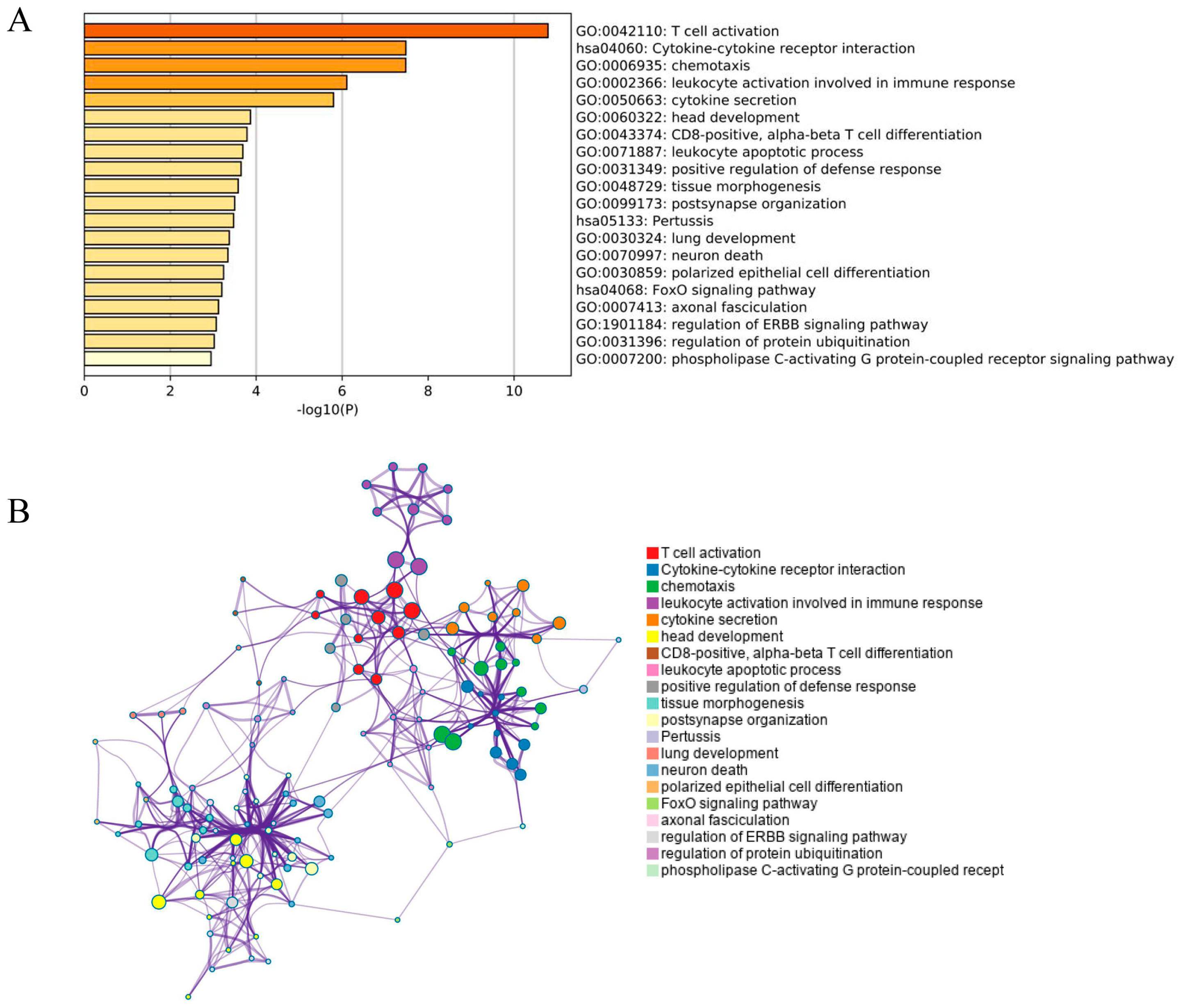
3.2. GO Analysis of DEGs
3.3. PPI Network Construction and Hub Gene Selection
3.4. Module Analysis
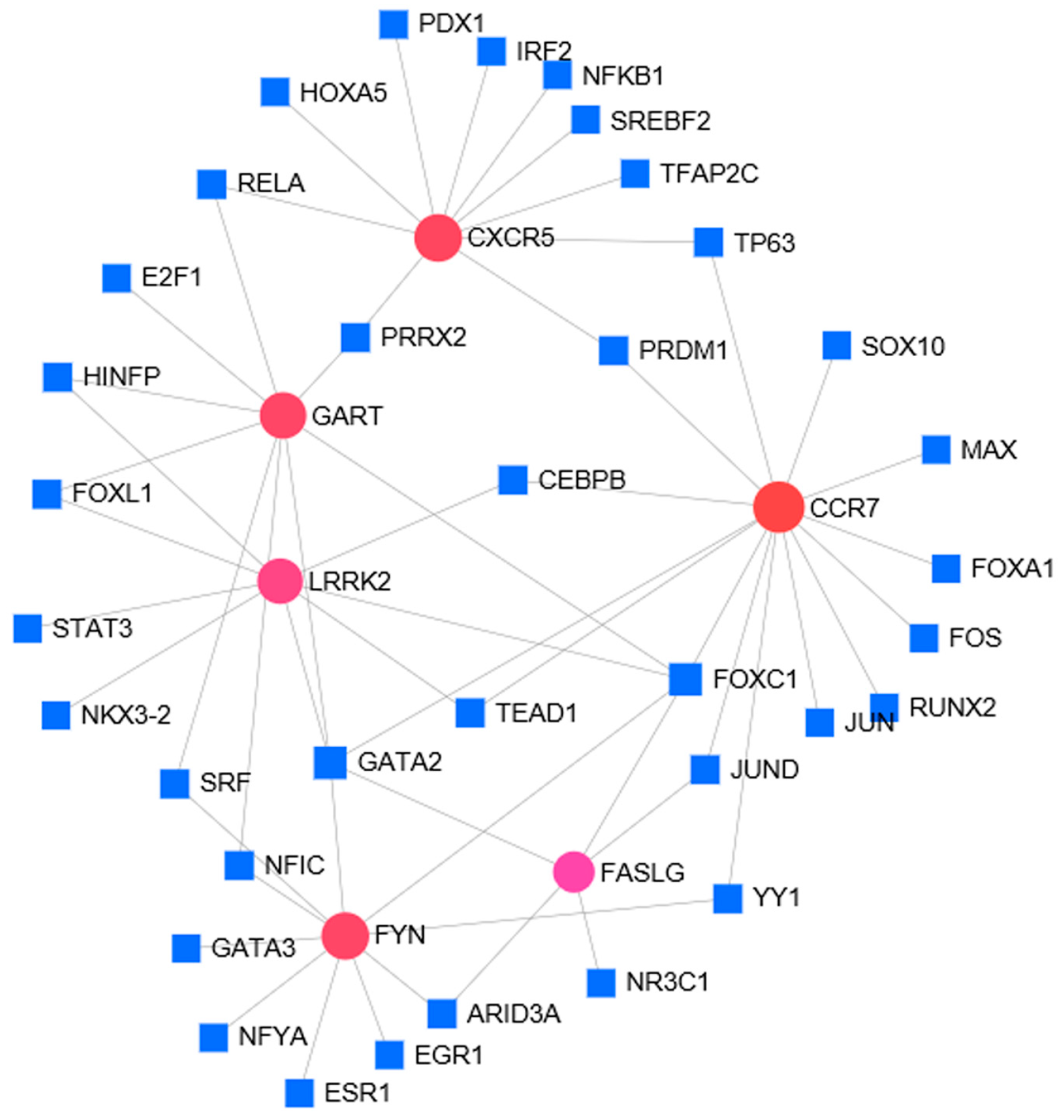
3.5. TF Regulatory Network Analysis of 6 Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laurens, A.H.; Joseph, P.C.; Birgitte, H.S.; Else, M.A.; Peter, A. Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. Eur. J. Immunol. 2015, 30, 3689–3698. [Google Scholar]
- Fan, S.; Zhou, G.; Shang, P.; Meng, L. Clinical Study of 660 Cases of Pulmonary Tuberculosis. Harbin Med. J. 2014, 34, 11–15. [Google Scholar]
- Siddiqi, K.; Lambert, M.-L.; Walley, J.D. Clinical diagnosis of smear-negative pulmonary tuberculosis in low-income countries: The current evidence. Lancet Infect. Dis. 2003, 3, 288–296. [Google Scholar] [CrossRef]
- Zhuang, Z.-G.; Zhang, J.-A.; Luo, H.-L.; Liu, G.-B.; Lu, Y.-B.; Ge, N.; Zheng, B.-Y.; Li, R.X.; Chen, C.; Wang, X.; et al. The circular RNA of peripheral blood mononuclear cells: Hsa_circ_0005836 as a new diagnostic biomarker and therapeutic target of active pulmonary tuberculosis. Mol. Immunol. 2017, 90, 264–272. [Google Scholar] [CrossRef]
- Roe, J.K.; Thomas, N.; Gil, E.; Best, K.; Tsaliki, E.; Morris-Jones, S.; Stafford, S.; Simpson, N.; Witt, K.D.; Chain, B.; et al. Blood transcriptomic diagnosis of pulmonary and extrapulmonary tuberculosis. JCI Insight 2016, 1, e87238. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Y.; Pei, Q.; Di, J.; Zhang, S. Serum concentrations of A disintegrin and metalloproteinase 9 (ADAM9) mRNA as a promising novel marker for the detection of pulmonary sarcoidosis. J. Int. Med. Res. 2013, 41, 1236–1241. [Google Scholar] [CrossRef] [Green Version]
- John, S.H.; Kenneth, J.; Gandhe, A.S. Host biomarkers of clinical relevance in tuberculosis: Review of gene and protein expression studies. Biomarkers 2011, 17, 1–8. [Google Scholar] [CrossRef]
- Dhanasekaran, S.; TB Trials Study Group; Jenum, S.; Stavrum, R.; Ritz, C.; Faurholt-Jepsen, D.; Kenneth, J.; Vaz, M.; Grewal, H.M.; Doherty, T.M. Identification of biomarkers for Mycobacterium tuberculosis infection and disease in BCG-vaccinated young children in Southern India. Genes Immun. 2013, 14, 356–364. [Google Scholar] [CrossRef]
- Füsun, S.; Pinar, Y. Serum CA-125: Biomarker of pulmonary tuberculosis activity and evaluation of response to treatment. Clinical & Investigative Medicine. Médecine Clin. Exp. 2012, 35, E223. [Google Scholar]
- Suzuki, Y.; Shirai, M.; Asada, K.; Miwa, S.; Karayama, M.; Nakamura, Y.; Inui, N.; Shirai, T.; Hayakawa, H.; Baba, S.; et al. Utility of Macrophage-activated Marker CD163 for Diagnosis and Prognosis in Pulmonary Tuberculosis. Ann. Am. Thorac. Soc. 2017, 14, 57–64. [Google Scholar] [CrossRef]
- Chowdhury, I.H.; Ahmed, A.M.; Choudhuri, S.; Sen, A.; Hazra, A.; Pal, N.K.; Bhattacharya, B.; Bahar, B. Alteration of serum inflammatory cytokines in active pulmonary tuberculosis following anti-tuberculosis drug therapy. Mol. Immunol. 2014, 62, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-R.; Wang, J.; Qiao, J.; Yi, Z.-J. Signature of circular RNAs in peripheral blood mononuclear cells from patients with active tuberculosis. J. Cell. Mol. Med. 2018, 23, 1917–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanunfre, K.A.; Leite, O.H.M.; Lopes, M.I.; Litvoc, M.; Ferreira, A.W. Enhancement of Diagnostic Efficiency by a Gamma Interferon Release Assay for Pulmonary Tuberculosis. Clin. Vaccine Immunol. 2008, 15, 1028–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.-B.; Zhang, W.-J.; Zou, L.; Huang, P.-J.; Sun, B.-J. Identification potential biomarkers in pulmonary tuberculosis and latent infection based on bioinformatics analysis. BMC Infect. Dis. 2016, 16, 500. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-W.; Wu, L.S.-H.; Huang, G.-M.; Huang, K.-Y.; Lee, T.-Y.; Weng, J.T.-Y. Gene expression profiling identifies candidate biomarkers for active and latent tuberculosis. BMC Bioinform. 2016, 17, S3. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Yang, Q.; Tang, Y.; Zhang, M.; Liu, H.; Zhang, G.; Deng, Q.; Huang, J.; Gao, Z.; Zhou, B.; et al. Increased Complement C1q Level Marks Active Disease in Human Tuberculosis. PLoS ONE 2014, 9, e92340. [Google Scholar] [CrossRef] [Green Version]
- Dumas, J.; Gargano, M.; Dancik, G.M. Abstract 5292: An online tool for biomarker analysis in Gene Expression Omnibus (GEO) datasets. Bioinform. Syst. Biol. 2016, 76, 5292. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.W.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; Von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012, 41, D808–D815. [Google Scholar] [CrossRef] [Green Version]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for Visualization and Analysis of Biological Networks. Adv. Struct. Saf. Stud. 2010, 696, 291–303. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.D.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uplekar, M.; Weil, D.; Lonnroth, K.; Jaramillo, E.; Lienhardt, C.; Dias, H.M.; Falzon, D.; Floyd, K.; Gargioni, G.; Getahun, H.; et al. WHO’s new end TB strategy. Lancet 2015, 385, 1799–1801. [Google Scholar] [CrossRef]
- Wang, C.; Liu, C.-M.; Wei, L.-L.; Shi, L.-Y.; Pan, Z.-F.; Mao, L.-G.; Wan, X.-C.; Ping, Z.-P.; Jiang, T.-T.; Chen, Z.-L.; et al. A Group of Novel Serum Diagnostic Biomarkers for Multidrug-Resistant Tuberculosis by iTRAQ-2D LC-MS/MS and Solexa Sequencing. Int. J. Boil. Sci. 2016, 12, 246–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Deweerd, N.A.; Stifter, S.A.; Liu, L.; Zhou, B.; Wang, W.; Zhou, Y.; Ying, B.; Hu, X.; Matthews, A.Y.; et al. A proline deletion in IFNAR1 impairs IFN-signaling and underlies increased resistance to tuberculosis in humans. Nat. Commun. 2018, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Jie, L.; Kumar, P.; Hong, G.S.; Leow, M.K.S.; Paleja, B.; Tsenova, L.; Kurepina, N.; Chen, J.; Zolezzi, F.; et al. Metformin as adjunct antituberculosis therapy. Sci. Transl. Med. 2014, 6, 263ra159. [Google Scholar] [CrossRef] [PubMed]
- Hart, R.P.; Herbst, S.; Peltier, J.; Rodgers, A.; Bilkei-Gorzo, O.; Fearns, A.; Dill, B.D.; Lee, H.; Flynn, R.; Cowley, S.A.; et al. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J. 2018, 37, e98694. [Google Scholar] [CrossRef]
- Mi, J.K.; Helen, C.W.; Michael, L.; Linda-Gail, B.; Gabriele, B.W.; Corneli, D.; Annalie, V.; Wei, W.; Fong-Fu, H.; Ursula, W.; et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2, 258–274. [Google Scholar]
- Slight, S.R.; Rangelmoreno, J.; Gopal, R.; Lin, Y.; Fallert, J.B.A.; Mehra, S.; Selman, M.; Becerril-Villanueva, E.; Baquera-Heredia, J.; Pavon, L.; et al. CXCR5⁺ T helper cells mediate protective immunity against tuberculosis. J. Clin. Investig. 2013, 123, 712–726. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, T.; Mogga, S.J.; Mfinanga, S.G.M.; Mørkve, O.; Sviland, L. Significance of Fas and Fas ligand in tuberculous lymphadenitis. Immunology 2005, 114, 255–262. [Google Scholar] [CrossRef]
- Lu, Y.-B.; Xiao, D.-Q.; Liang, K.-D.; Zhang, J.-A.; Wang, W.-D.; Yu, S.-Y.; Zheng, B.-Y.; Gao, Y.-C.; Dai, Y.-C.; Jia, Y.; et al. Profiling dendritic cell subsets in the patients with active pulmonary tuberculosis. Mol. Immunol. 2017, 91, 86–96. [Google Scholar] [CrossRef]
- Gilding, L.N.; Somervaille, T.C.P. The Diverse Consequences of FOXC1 Deregulation in Cancer. Cancers 2019, 11, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquet, M.; Bellanné-Chantelot, C.; Tavitian, S.; Prade, N.; Beaupain, B.; LaRochelle, O.; Petit, A.; Rohrlich, P.S.; Ferrand, C.; Neste, E.V.D.; et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood 2013, 121, 822–829. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TFs | Genes | Count |
|---|---|---|
| FOXC1 | LRRK2, FYN, GART, CCR7, CXCR5, and FASLG | 6 |
| GATA2 | FASLG, GART, LRRK2, CCR7 | 4 |
| PRDM1 | CXCR5, CCR7 | 2 |
| TP63 | CXCR5, CCR7 | 2 |
| PRRX2 | CXCR5, GART | 2 |
| RELA | CXCR5, GART | 2 |
| YY1 | FYN, CCR7 | 2 |
| NFIC | FYN, GART | 2 |
| SRF | FYN, GART | 2 |
| CEBPB | LRRK2, CCR7 | 2 |
| TEAD1 | LRRK2, CCR7 | 2 |
| JUND | FASLG, CCR7 | 2 |
| FOXL1 | LRRK2, GART | 2 |
| HINFP | LRRK2, GART | 2 |
| ARID3A | FYN, FASLG | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, L.; Chao, X.; Teng, T.; Li, Q.; Xie, J. Identification of Potential Biomarkers and Related Transcription Factors in Peripheral Blood of Tuberculosis Patients. Int. J. Environ. Res. Public Health 2020, 17, 6993. https://doi.org/10.3390/ijerph17196993
Xie L, Chao X, Teng T, Li Q, Xie J. Identification of Potential Biomarkers and Related Transcription Factors in Peripheral Blood of Tuberculosis Patients. International Journal of Environmental Research and Public Health. 2020; 17(19):6993. https://doi.org/10.3390/ijerph17196993
Chicago/Turabian StyleXie, Longxiang, Xiaoyu Chao, Tieshan Teng, Qiming Li, and Jianping Xie. 2020. "Identification of Potential Biomarkers and Related Transcription Factors in Peripheral Blood of Tuberculosis Patients" International Journal of Environmental Research and Public Health 17, no. 19: 6993. https://doi.org/10.3390/ijerph17196993