Environmental Exposures during Puberty: Window of Breast Cancer Risk and Epigenetic Damage

 , , , ,
, , , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
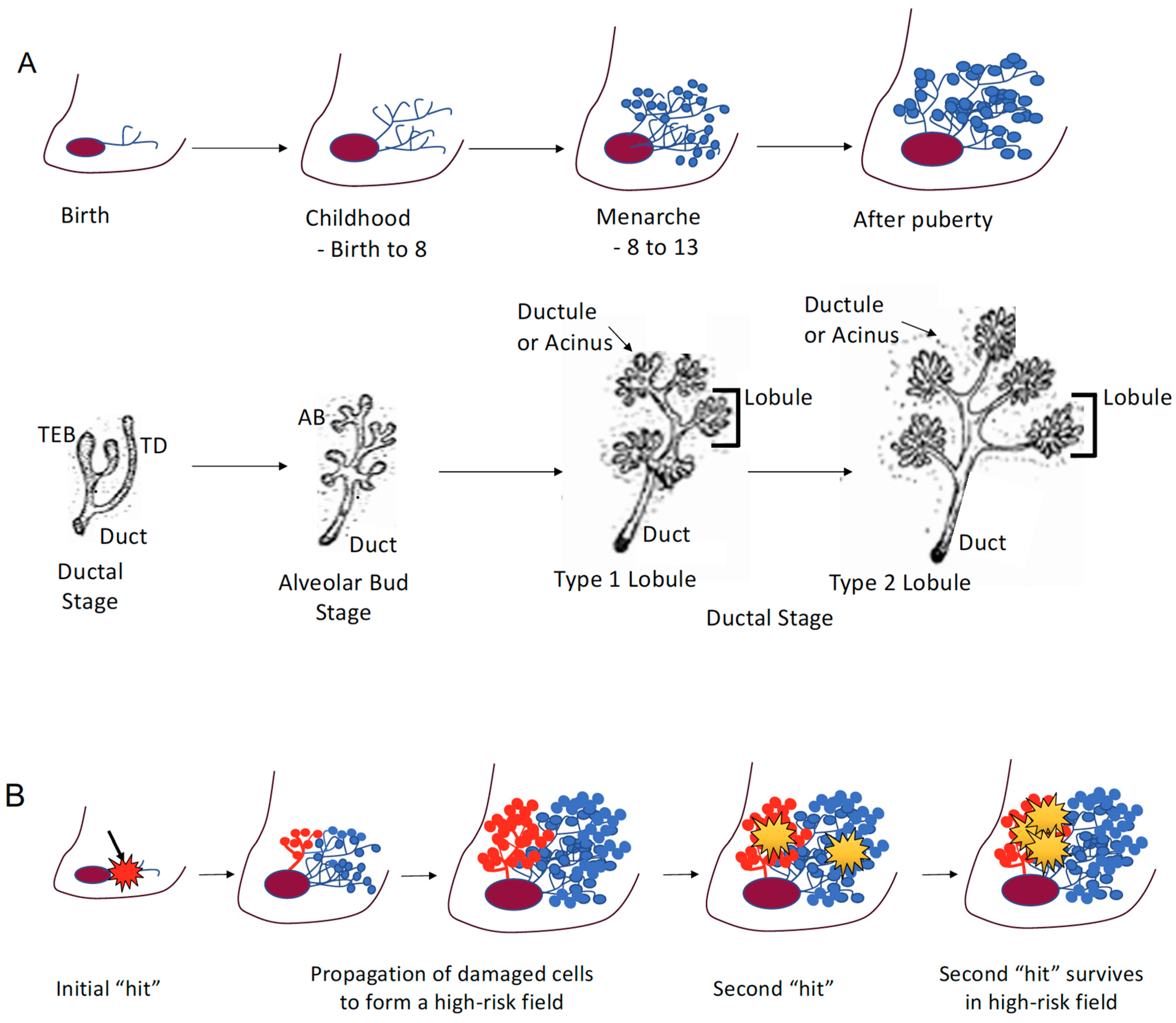
2. Puberty and Susceptibility
2.1. Puberty
2.2. High-Risk Field
2.3. Impact of Delayed Pregnancies

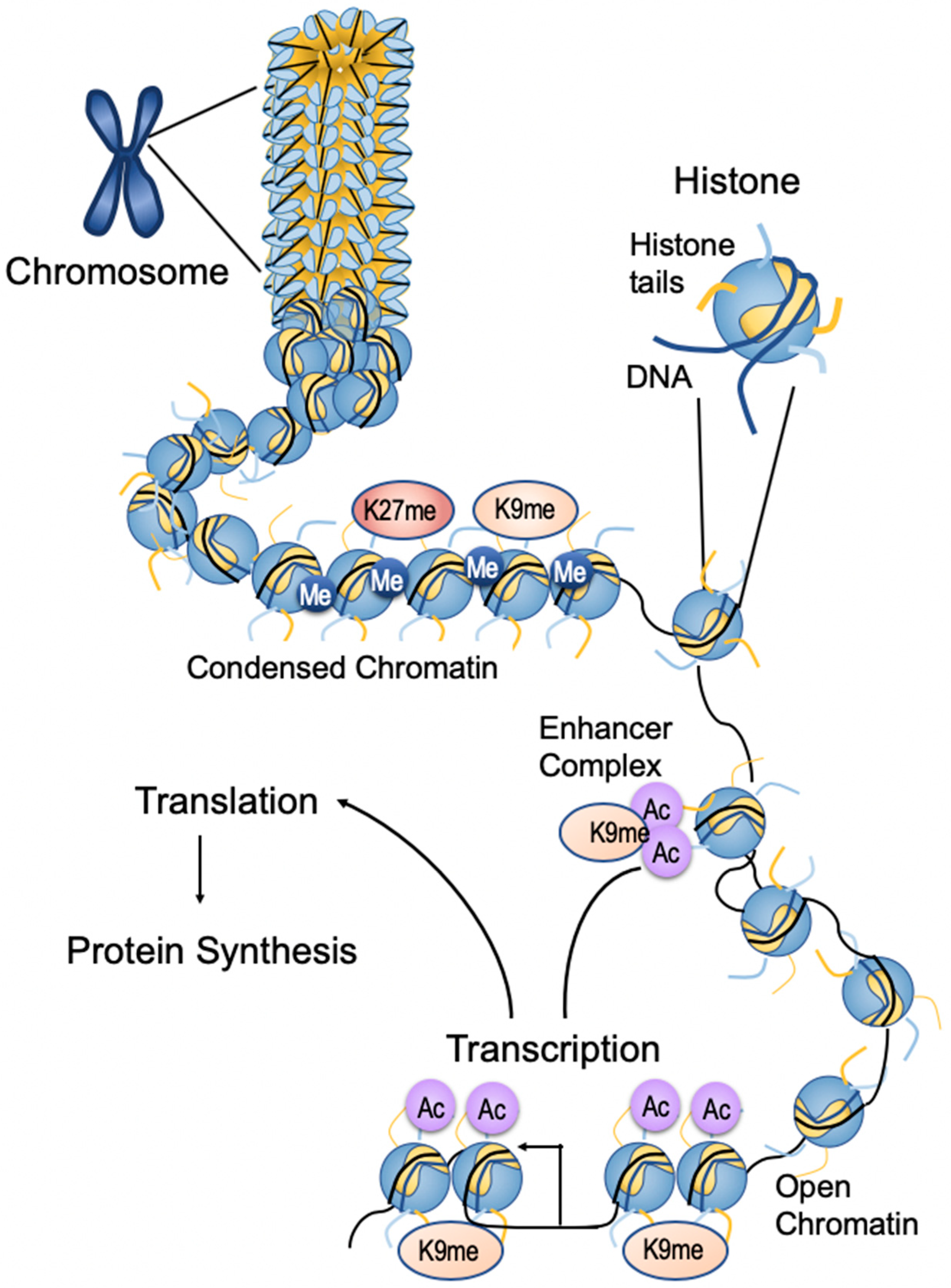
3. Epigenetics
3.1. Epigenetic Changes Associated with Breast Cancer
3.2. Writers/Erasers
3.3. Histone Modification
3.4. LncRNAs
3.5. Imprinting
3.6. Gene-Epigenenome Interactions

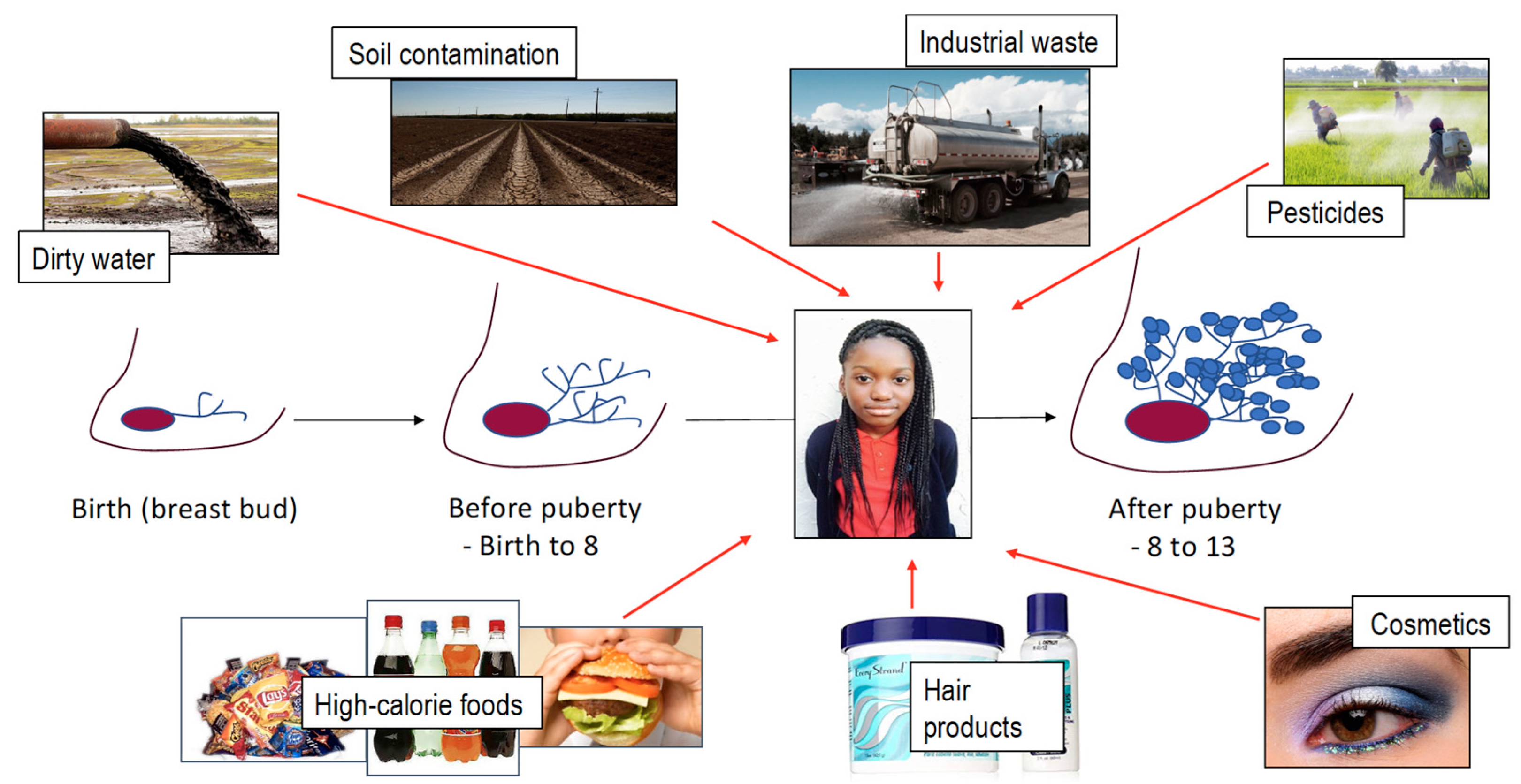
4. Exposures
4.1. Water
4.2. Air Pollution
4.3. Cosmetics
4.3.1. Formaldehyde
4.3.2. Aluminum Salts
4.3.3. Endocrine Disrupting Chemicals (EDC)
5. Nutrition, Obesity, Risk
5.1. Obesity, Breast Cancer, Controversy
5.2. Energy-Dense Foods and Risk
5.3. Diet Soda
6. Stress
7. Neighborhoods
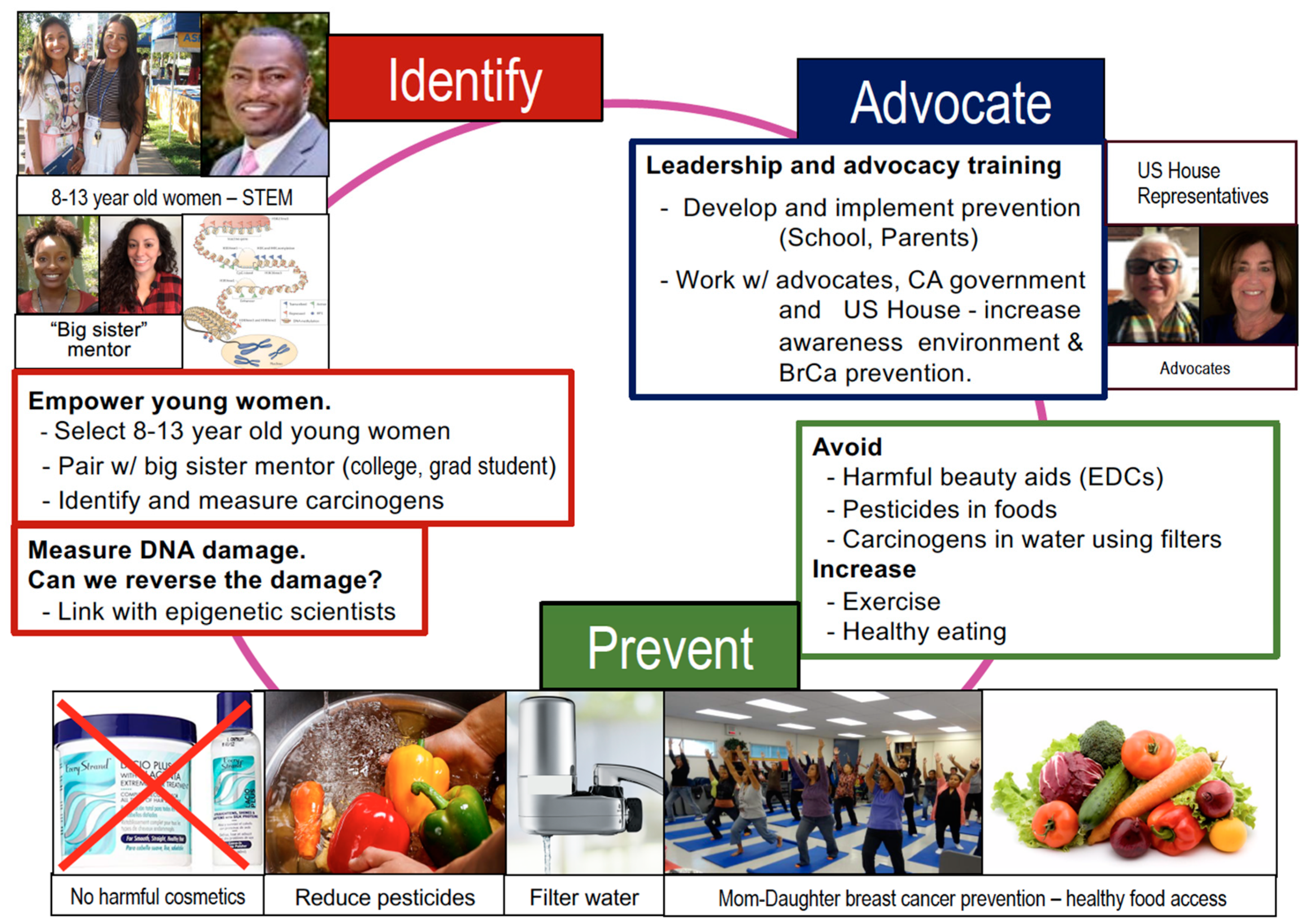
8. Conclusions—Call for Advocacy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deng, G.; Lu, Y.; Zlotnikov, G.; Thor, A.D.; Smith, H.S. Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science 1996, 274, 2057–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, J.; Tay, L.K.; Russo, I.H. Differentiation of the mammary gland and susceptibility to carcinogenesis. Breast Cancer Res. Treat. 1982, 2, 5–73. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Wilgus, G.; Russo, I.H. Susceptibility of the mammary gland to carcinogenesis: I Differentiation of the mammary gland as determinant of tumor incidence and type of lesion. Am. J. Pathol. 1979, 96, 721–736. [Google Scholar] [PubMed]
- Terry, M.B.; Michels, K.B.; Brody, J.G.; Byrne, C.; Chen, S.; Jerry, D.J.; Malecki, K.M.C.; Martin, M.B.; Miller, R.L.; Neuhausen, S.L.; et al. Environmental exposures during windows of susceptibility for breast cancer: A framework for prevention research. Breast Cancer Res. 2019, 21, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilakivi-Clarke, L. Maternal exposure to diethylstilbestrol during pregnancy and increased breast cancer risk in daughters. Breast Cancer Res. 2014, 16, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechuta, S.; Paneth, N.; Velie, E.M. Pregnancy characteristics and maternal breast cancer risk: A review of the epidemiologic literature. Cancer Causes Control 2010, 21, 967–989. [Google Scholar] [CrossRef]
- Nichols, H.B.; Schoemaker, M.J.; Cai, J.; Xu, J.; Wright, L.B.; Brook, M.N.; Jones, M.E.; Adami, H.O.; Baglietto, L.; Bertrand, K.A.; et al. Breast Cancer Risk After Recent Childbirth: A Pooled Analysis of 15 Prospective Studies. Ann. Intern. Med. 2019, 170, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Frech, M.S.; Halama, E.D.; Tilli, M.T.; Singh, B.; Gunther, E.J.; Chodosh, L.A.; Flaws, J.A.; Furth, P.A. Deregulated estrogen receptor alpha expression in mammary epithelial cells of transgenic mice results in the development of ductal carcinoma in situ. Cancer Res. 2005, 65, 681–685. [Google Scholar]
- Jones, L.P.; Tilli, M.T.; Assefnia, S.; Torre, K.; Halama, E.D.; Parrish, A.; Rosen, E.M.; Furth, P.A. Activation of estrogen signaling pathways collaborates with loss of Brca1 to promote development of ERalpha-negative and ERalpha-positive mammary preneoplasia and cancer. Oncogene 2008, 27, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Robison, L.L.; Oberlin, O.; Greenberg, M.; Bunin, G.; Fossati-Bellani, F.; Meadows, A.T. Breast cancer and other second neoplasms after childhood Hodgkin’s disease. N. Engl. J. Med. 1996, 334, 745–751. [Google Scholar] [CrossRef]
- Land, C.E. Studies of cancer and radiation dose among atomic bomb survivors. The example of breast cancer. JAMA 1995, 274, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Cohn, B.A.; Wolff, M.S.; Cirillo, P.M.; Sholtz, R.I. DDT and breast cancer in young women: New data on the significance of age at exposure. Environ. Health Perspect. 2007, 115, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Cohn, B.A.; Cirillo, P.M.; Terry, M.B. DDT and Breast Cancer: Prospective Study of Induction Time and Susceptibility Windows. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Moschetta, M.; Tatsi, K.; Tsiouris, A.K.; Pavlidis, N. A review on pregnancy complicated by ovarian epithelial and non-epithelial malignant tumors: Diagnostic and therapeutic perspectives. J. Adv. Res. 2018, 12, 1–9. [Google Scholar] [CrossRef]
- Cardiff, R.D.; Jindal, S.; Treuting, P.M.; Going, J.J.; Gusterson, B.A.; Thompson, H.J. Mammary Gland. In Comparative Anatomy and Histology: A Mouse, Rat, and Human Atlas, 2nd ed.; Treuting, P.M., Dintzis, S.M., Montine, K.S., Eds.; Academic Press: London, UK, 2018; pp. 487–509. [Google Scholar]
- Nilsson, E.E.; Skinner, M.K. Environmentally induced epigenetic transgenerational inheritance of disease susceptibility. Transl. Res. 2015, 165, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015, 58, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Keating, S.T.; Plutzky, J.; El-Osta, A. Epigenetic Changes in Diabetes and Cardiovascular Risk. Circ. Res. 2016, 118, 1706–1722. [Google Scholar] [CrossRef] [Green Version]
- Pinney, S.E.; Simmons, R.A. Metabolic programming, epigenetics, and gestational diabetes mellitus. Curr. Diab. Rep. 2012, 12, 67–74. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Cooke, D.; Neille, C.; Sadauskaite, A.; Moschetta, M.; Zakynthinakis-Kyriakou, N.; Pavlidis, N. PARP Inhibitors in Ovarian Cancer: The Route to “Ithaca”. Diagnostics 2019, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.S. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol. Oncol. 2015, 137, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Hampton, D.D.; Jirtle, R.L. Imprinting evolution and human health. Mamm. Genome. 2009, 20, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Moore, G.E. The role of imprinted genes in humans. Mol. Aspects Med. 2013, 34, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Hoyo, C. Sculpting Our Future: Environmental nudging of the imprintome. In Environmental Epigenomics in Health and Disease; Jirtle, R.L., Tyson, F.L., Eds.; Springer: Berlin, Germany, 2013; pp. 51–73. [Google Scholar]
- Soubry, A.; Hoyo, C.; Jirtle, R.L.; Murphy, S.K. A paternal environmental legacy: Evidence for epigenetic inheritance through the male germ line. Bioessays 2014, 36, 359–371. [Google Scholar] [CrossRef]
- Murphy, S.K.; Huang, Z.; Hoyo, C. Differentially methylated regions of imprinted genes in prenatal, perinatal and postnatal human tissues. PLoS ONE 2012, 7, e40924. [Google Scholar] [CrossRef] [Green Version]
- Hoyo, C.; Daltveit, A.K.; Iversen, E.; Benjamin-Neelon, S.E.; Fuemmeler, B.; Schildkraut, J.; Murtha, A.P.; Overcash, F.; Vidal, A.C.; Wang, F.; et al. Erythrocyte folate concentrations, CpG methylation at genomically imprinted domains, and birth weight in a multiethnic newborn cohort. Epigenetics 2014, 9, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Vidal, A.C.; Neelon, S.E.B.; Liu, Y.; Tuli, A.M.; Fuemmeler, B.F.; Hoyo, C.; Murtha, A.P.; Huang, Z.; Schildkraut, J.; Overcash, F.; et al. Maternal stress, preterm birth, and DNA methylation at imprint regulatory sequences in humans. Genet. Epigenet. 2014, 6, 37–44. [Google Scholar] [CrossRef]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Varrault, A.; Gueydan, C.; Delalbre, A.; Bellmann, A.; Houssami, S.; Aknin, C.; Severac, D.; Chotard, L.; Kahli, M.; Le Digarcher, A.; et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 2006, 11, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Small, K.S.; Hedman, A.K.; Grundberg, E.; Nica, A.C.; Thorleifsson, G.; Kong, A.; Thorsteindottir, U.; Shin, S.Y.; Richards, H.B.; GIANT Consortium; et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 2011, 43, 561–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.C.; Boileau, P.; Le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child health, developmental plasticity, and epigenetic programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar] [CrossRef]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef]
- WHO. Water Quality and Health Strategy 2013–2020. 2012. Available online: https://www.who.int/water_sanitation_health/publications/water_quality_strategy/en/ (accessed on 4 October 2019).
- Sharp, K.A. Water: Structure and properties. In Encyclopedia of Life Sciences; John Wiley & Sons: Hoboken, NJ, USA, 2001; Available online: http://crystal.med.upenn.edu/sharp-lab-pdfs/sharp_EncLifeSci.pdf (accessed on 4 October 2019).
- Lack of Access to Safe Water Perpetuates Poverty. Available online: https://news.un.org/en/story/2010/09/351992-lack-access-safe-water-perpetuates-poverty-ban-cautions (accessed on 4 October 2019).
- Hanna-Attisha, M.; LaChance, J.; Sadler, R.C.; Champney Schnepp, A. Elevated Blood Lead Levels in Children Associated with the Flint Drinking Water Crisis: A Spatial Analysis of Risk and Public Health Response. Am. J. Public Health 2016, 106, 283–290. [Google Scholar] [CrossRef]
- Public Health Impacts of Gaza’s Water Crisis. Available online: https://www.rand.org/pubs/research_reports/RR2515.html (accessed on 14 November 2019).
- Ganguly, K.; Levanen, B.; Palmberg, L.; Akesson, A.; Linden, A. Cadmium in tobacco smokers: A neglected link to lung disease? Eur. Respir. Rev. 2018, 27. [Google Scholar] [CrossRef]
- Schweitzer, L.; Noblet, J. Water Contamination and Pollution. In Green Chemistry: An Inclusive Approach; Torok, B., Ed.; Elsevier: Cambridge, MA, USA, 2017; pp. 261–290. [Google Scholar]
- Jadoon SaM, A. DNA Damage by Heavy Metals in Animals and Human Beings: An overview. Biochem. Pharmacol. 2017. [Google Scholar] [CrossRef]
- Langie, S.A.; Koppen, G.; Desaulniers, D.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Azqueta, A.; Bisson, W.H.; Brown, D.G.; Brunborg, G.; et al. Causes of genome instability: The effect of low dose chemical exposures in modern society. Carcinogenesis 2015, 36, S61–S88. [Google Scholar] [CrossRef]
- Park, S.S.; Skaar, D.A.; Jirtle, R.L.; Hoyo, C. Epigenetics, obesity and early-life cadmium or lead exposure. Epigenomics 2017, 9, 57–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowley, M.; Skaar, D.A.; Jima, D.D.; Maguire, R.L.; Hudson, K.M.; Park, S.S.; Sorrow, P.; Hoyo, C. Effects of Cadmium Exposure on DNA Methylation at Imprinting Control Regions and Genome-Wide in Mothers and Newborn Children. Environ. Health Perspect. 2018, 126, 037003. [Google Scholar] [CrossRef] [PubMed]
- Everson, T.M.; Punshon, T.; Jackson, B.P.; Hao, K.; Lambertini, L.; Chen, J.; Karagas, M.R.; Marsit, C.J. Cadmium-Associated Differential Methylation throughout the Placental Genome: Epigenome-Wide Association Study of Two, U.S. Birth Cohorts. Environ. Health Perspect. 2018, 126, 017010. [Google Scholar] [CrossRef] [Green Version]
- Carcinogenic Ingredients in Your Personal Care Products. Available online: https://www.healthline.com/health/carcinogenic-ingredients-your-personal-care-products#common-chemicals (accessed on 4 October 2019).
- Sahay, D.; Terry, M.B.; Miller, R. Is breast cancer a result of epigenetic responses to traffic-related air pollution? A review of the latest evidence. Epigenomics 2019, 11, 701–714. [Google Scholar] [CrossRef]
- Rider, C.F.; Carlsten, C. Air pollution and DNA methylation: Effects of exposure in humans. Clin. Epigenet. 2019, 11, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmuel, S.; White, A.J.; Sandler, D.P. Residential exposure to vehicular traffic-related air pollution during childhood and breast cancer risk. Environ. Res. 2017, 159, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Hurley, S.; Nelson, D.O.; Hertz, A.; Reynolds, P. Hazardous air pollutants and breast cancer risk in California teachers: A cohort study. Environ. Health 2015, 14, 14. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; Taylor, K.E. Concealing Danger: How the Regulation of Cosmetics in the United States Put Consumersat Risk; Berkeley Electronic Press: Berkeley, CA, USA, 2011; Available online: https://ir.lawnet.fordham.edu/cgi/viewcontent.cgi?article=1696&context=elr (accessed on 4 October 2019).
- Califf, R.M.; McCall, J.; Mark, D.B. Cosmetics, Regulations, and the Public Health: Understanding the Safety of Medical and Other Products. JAMA Intern. Med. 2017, 177, 1080–1082. [Google Scholar] [CrossRef]
- Jacob, S.L.; Cornell, E.; Kwa, M.; Funk, W.E.; Xu, S. Cosmetics and Cancer: Adverse Event Reports Submitted to the Food and Drug Administration. JNCI Cancer Spectr. 2018, 2. [Google Scholar] [CrossRef]
- Bray, K. Endocrine-Disrupting Chemicals: Which Common Chemicals Have Been Linked to Cancer and Reproductive Abnormalities? Available online: https://www.choice.com.au/health-and-body/beauty-and-personal-care/skin-care-and-cosmetics/articles/endocrine-disrupting-chemicals (accessed on 4 October 2019).
- Konduracka, E.; Krzemieniecki, K.; Gajos, G. Relationship between everyday use cosmetics and female breast cancer. Pol. Arch. Med. Wewn. 2014, 124, 264–269. [Google Scholar] [CrossRef]
- Macon, M.B.; Fenton, S.E. Endocrine disruptors and the breast: Early life effects and later life disease. J. Mammary Gland Biol. Neoplasia 2013, 18, 43–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watnick, V.J. The Missing Link: U.S. regulation of consumer cosmetics products to protect human health and the environment. Pace Environ. Law Rev. 2014, 31, 595–650. [Google Scholar]
- Marchese, M. Parabens and Breast Cancer: Parabens are preservatives used in a wide range of topical cosmetic products. Nat. Med. J. 2010, 2, 1–3. [Google Scholar]
- Darbre, P.D.; Aljarrah, A.; Miller, W.R.; Coldham, N.G.; Sauer, M.J.; Pope, G.S. Concentrations of parabens in human breast tumours. J. Appl. Toxicol. 2004, 24, 5–13. [Google Scholar] [CrossRef]
- Reeves, K.W.; Díaz Santana, M.; Manson, J.E.; Hankinson, S.E.; Zoeller, R.T.; Bigelow, C.; Sturgeon, S.R.; Spiegelman, D.; Tinker, L.; Luo, J.; et al. Urinary Phthalate Biomarker Concentrations and Postmenopausal Breast Cancer Risk. J. Natl. Cancer Inst. 2019, 111, 1059–1067. [Google Scholar] [CrossRef]
- Cirillo, T.; Fasano, E.; Castaldi, E.; Montuori, P.; Amodio Cocchieri, R. Children’s exposure to Di(2-ethylhexyl)phthalate and dibutylphthalate plasticizers from school meals. J. Agric. Food Chem. 2011, 59, 10532–10538. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.Y.; Huang, P.C.; Lee, C.C.; Wu, M.H.; Lin, S.J. Phthalate exposure in girls during early puberty. J. Pediatr. Endocrinol. Metab. 2009, 22, 69–77. [Google Scholar] [CrossRef]
- Kay, V.R.; Chambers, C.; Foster, W.G. Reproductive and developmental effects of phthalate diesters in females. Crit. Rev. Toxicol. 2013, 43, 200–219. [Google Scholar] [CrossRef] [Green Version]
- About Cancer: Prevention, Risk, and Diet. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/diet (accessed on 4 October 2019).
- Harris, H.R.; Willett, W.C.; Vaidya, R.L.; Michels, K.B. Adolescent dietary patterns and premenopausal breast cancer incidence. Carcinogenesis 2016, 37, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Farvid, M.S.; Eliassen, A.H.; Cho, E.; Liao, X.; Chen, W.Y.; Willett, W.C. Dietary Fiber Intake in Young Adults and Breast Cancer Risk. Pediatrics 2016, 137, e20151226. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Chen, Y.; Ma, S.; Zheng, R.; Zhao, P.; Zhang, L.; Liu, Y.; Yu, Q.; Deng, Q.; Zhang, K. Dietary fibre intake and risk of breast cancer: A systematic review and meta-analysis of epidemiological studies. Oncotarget 2016, 7, 80980–80989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, S.; Macintyre, S. “Food deserts”—Evidence and assumption in health policy making. BMJ 2002, 325, 436–438. [Google Scholar] [CrossRef]
- Dietze, E.C.; Chavez, T.A.; Seewaldt, V.L. Obesity and Triple-Negative Breast Cancer: Disparities, Controversies, and Biology. Am. J. Pathol. 2018, 188, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, T.L.; Wilson, K.E.; Heymsfield, S.B. Dual energy X-Ray absorptiometry body composition reference values from NHANES. PLoS ONE 2009, 4, e7038. [Google Scholar] [CrossRef]
- Heo, M.; Kabat, G.C.; Gallagher, D.; Heymsfield, S.B.; Rohan, T.E. Optimal scaling of weight and waist circumference to height for maximal association with DXA-measured total body fat mass by sex, age and race/ethnicity. Int. J. Obes. 2013, 37, 1154–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Heo, M.; Lee, R.C.; Kotler, D.P.; Withers, R.T.; Heymsfield, S.B. Muscularity in adult humans: Proportion of adipose tissue-free body mass as skeletal muscle. Am. J. Hum. Biol. 2001, 13, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Shen, W.; Heo, M.; Gallagher, D.; Wang, Z.; Sardinha, L.B.; Heymsfield, S.B. Ethnicity-related skeletal muscle differences across the lifespan. Am. J. Hum. Biol. 2010, 22, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Kwan, M.L.; John, E.M.; Caan, B.J.; Lee, V.S.; Bernstein, L.; Cheng, I.; Gomez, S.L.; Henderson, B.E.; Keegan, T.H.; Kurian, A.W.; et al. Obesity and mortality after breast cancer by race/ethnicity: The California Breast Cancer Survivorship Consortium. Am. J. Epidemiol. 2014, 179, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Chiu, M.; Austin, P.C.; Manuel, D.G.; Shah, B.R.; Tu, J.V. Deriving ethnic-specific BMI cutoff points for assessing diabetes risk. Diabetes Care 2011, 34, 1741–1748. [Google Scholar] [CrossRef] [Green Version]
- Chandran, U.; McCann, S.E.; Zirpoli, G.; Gong, Z.; Lin, Y.; Hong, C.C.; Ciupak, G.; Pawlish, K.; Ambrosone, C.B.; Bandera, E.V. Intake of energy-dense foods, fast foods, sugary drinks, and breast cancer risk in African American and European American women. Nutr. Cancer 2014, 66, 1187–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, V.S.; Li, Y.; Pan, A.; De Koning, L.; Schernhammer, E.; Willett, W.C.; Hu, F.B. Long-Term Consumption of Sugar-Sweetened and Artificially Sweetened Beverages and Risk of Mortality in US Adults. Circulation 2019, 139, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Sylvetsky, A.C.; Figueroa, J.; Zimmerman, T.; Swithers, S.E.; Welsh, J.A. Consumption of low-calorie sweetened beverages is associated with higher total energy and sugar intake among children, NHANES 2011–2016. Pediatr. Obes. 2019, 14, e12535. [Google Scholar] [CrossRef]
- Ruiz-Ojeda, F.J.; Plaza-Diaz, J.; Saez-Lara, M.J.; Gil, A. Effects of Sweeteners on the Gut Microbiota: A Review of Experimental Studies and Clinical Trials. Adv. Nutr. 2019, 10, S31–S48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, S. Developmental determinants of sensitivity and resistance to stress. Psychoneuroendocrinology 2005, 30, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, L.L.; Shattuck, T.T.; Tyrka, A.R.; Geracioti, T.D.; Price, L.H. Effect of childhood physical abuse on cortisol stress response. Psychopharmacology 2011, 214, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heim, C.; Ehlert, U.; Hellhammer, D.H. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology 2000, 25, 1–35. [Google Scholar] [CrossRef]
- Parker, K.J.; Schatzberg, A.F.; Lyons, D.M. Neuroendocrine aspects of hypercortisolism in major depression. Horm. Behav. 2003, 43, 60–66. [Google Scholar] [CrossRef]
- Hoffmann, A.; Spengler, D. The lasting legacy of social stress on the epigenome of the hypothalamic–pituitary–adrenal axis. Epigenomics 2012, 4, 431–444. [Google Scholar] [CrossRef]
- Bockmühl, Y.; Patchev, A.V.; Madejska, A.; Hoffmann, A.; Sousa, J.C.; Sousa, N.; Holsboer, F.; Almeida, O.F.; Spengler, D. Methylation at the CpG island shore region upregulates Nr3c1 promoter activity after early-life stress. Epigenetics 2015, 10, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Where You Live Affects How Long You Live. Available online: https://www.rwjf.org/en/library/interactives/whereyouliveaffectshowlongyoulive.html (accessed on 6 October 2019).
- Arias, E.; Escobedo, L.A.; Kennedy, J.; Fu, C.; Cisewki, J. US Small-Area Life Expectancy Estimates Project: Methodology and Results Summary. 2018. Available online: https://www.cdc.gov/nchs/data/series/sr_02/sr02_181.pdf (accessed on 14 November 2019).
- Maantay, J. Mapping environmental injustices: Pitfalls and potential of geographic information systems in assessing environmental health and equity. Environ. Health Perspect. 2002, 110, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Maantay, J.; Maroko, A. Mapping Urban Risk: Flood Hazards, Race, & Environmental Justice In New York. Appl. Geogr. 2009, 29, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.M.; Murray, R.T.; Jiang, C.; Dalemarre, L.; Burwell-Naney, K.; Fraser-Rahim, H. Environmental Justice Radar: A Tool for Community-Based Mapping to Increase Environmental Awareness and Participatory Decision Making. Prog. Community Health Partnersh. 2015, 9, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Sadd, J.L.; Pastor, M.; Morello-Frosch, R.; Scoggins, J.; Jesdale, B. Playing it safe: Assessing cumulative impact and social vulnerability through an environmental justice screening method in the South Coast Air Basin, California. Int. J. Environ. Res. Public Health 2011, 8, 1441–1459. [Google Scholar] [CrossRef] [PubMed]
- Food Desert Atlas. Available online: https://www.ers.usda.gov/data-products/food-access-research-atlas/go-to-the-atlas.aspx (accessed on 6 October 2019).
- Jankowska, M.M.; Yang, J.A.; Block, J.; Baer, R.J.; Jelliffe-Pawlowski, L.L.; Flores, S.; Pacheco-Warner, T.; Costantino, A.; Fuchs, J.; Chambers, C.D.; et al. An Online Geographic Data Visualization Tool to Relate Preterm Births to Environmental Factors. Prev. Chronic Dis. 2019, 16, 180498. [Google Scholar] [CrossRef] [Green Version]
- Hekler, E.B.; Klasnja, P.; Chevance, G.; Golaszewski, N.M.; Lewis, D.; Sim, I. Why we need a small data paradigm. BMC Med. 2019, 17, 133. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Jiang, B.; Zhu, P.; Geng, X.; Liu, Z.; Cui, L.; Yang, L. Associations between maternal weekly air pollutant exposures and low birth weight: A distributed lag non-linear model. Environ. Res. Lett. 2018, 13, 024023. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natarajan, R.; Aljaber, D.; Au, D.; Thai, C.; Sanchez, A.; Nunez, A.; Resto, C.; Chavez, T.; Jankowska, M.M.; Benmarhnia, T.; et al. Environmental Exposures during Puberty: Window of Breast Cancer Risk and Epigenetic Damage. Int. J. Environ. Res. Public Health 2020, 17, 493. https://doi.org/10.3390/ijerph17020493
Natarajan R, Aljaber D, Au D, Thai C, Sanchez A, Nunez A, Resto C, Chavez T, Jankowska MM, Benmarhnia T, et al. Environmental Exposures during Puberty: Window of Breast Cancer Risk and Epigenetic Damage. International Journal of Environmental Research and Public Health. 2020; 17(2):493. https://doi.org/10.3390/ijerph17020493
Chicago/Turabian StyleNatarajan, Rama, Dana Aljaber, Dawn Au, Christine Thai, Angelica Sanchez, Alan Nunez, Cristal Resto, Tanya Chavez, Marta M. Jankowska, Tarik Benmarhnia, and et al. 2020. "Environmental Exposures during Puberty: Window of Breast Cancer Risk and Epigenetic Damage" International Journal of Environmental Research and Public Health 17, no. 2: 493. https://doi.org/10.3390/ijerph17020493
APA StyleNatarajan, R., Aljaber, D., Au, D., Thai, C., Sanchez, A., Nunez, A., Resto, C., Chavez, T., Jankowska, M. M., Benmarhnia, T., Yang, J. -A., Jones, V., Tomsic, J., McCune, J. S., Sistrunk, C., Doan, S., Serrano, M., Cardiff, R. D., Dietze, E. C., & Seewaldt, V. L. (2020). Environmental Exposures during Puberty: Window of Breast Cancer Risk and Epigenetic Damage. International Journal of Environmental Research and Public Health, 17(2), 493. https://doi.org/10.3390/ijerph17020493