Identification of Potentially Therapeutic Target Genes of Hepatocellular Carcinoma

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Selection
2.2. Differential Expression Analysis of the Selected Gene Expression Profiles
2.3. GO (Gene Ontology) and KEGG (Kyoto Encyclopedia Genes and Genomes) Enrichment Analysis of Common DEGs
2.4. PPI Network Construction and Identification of Hub Genes
2.5. Validation of HCC Hub Gene Expression via Oncomine Database
2.6. Survival Analysis
3. Results
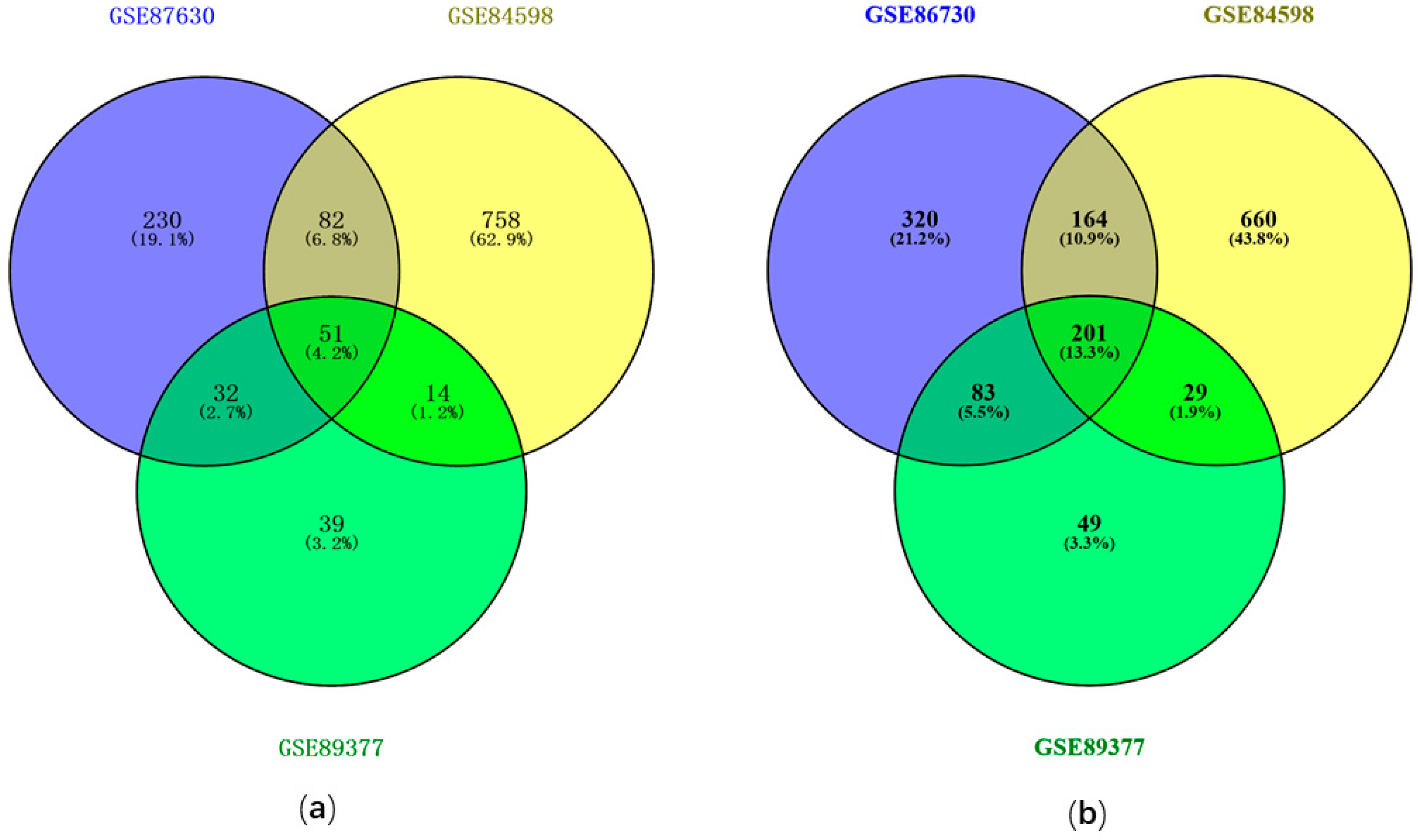
3.1. Identification of DEGs with GEO2R
3.2. GO and KEGG Enrichment Analysis
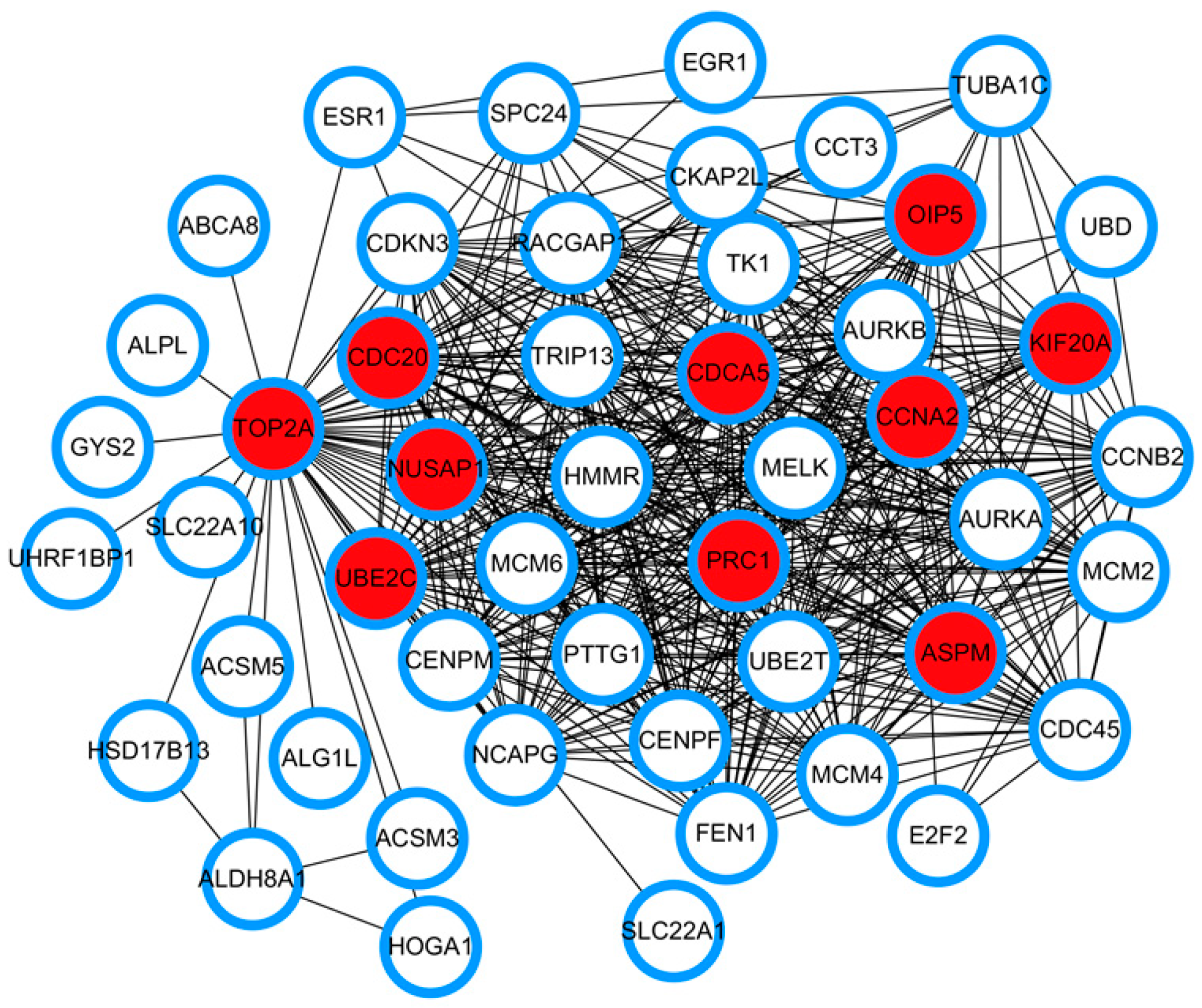
3.3. PPI Network Construction and Identification of Hub Genes Based on Network Topological Analysis
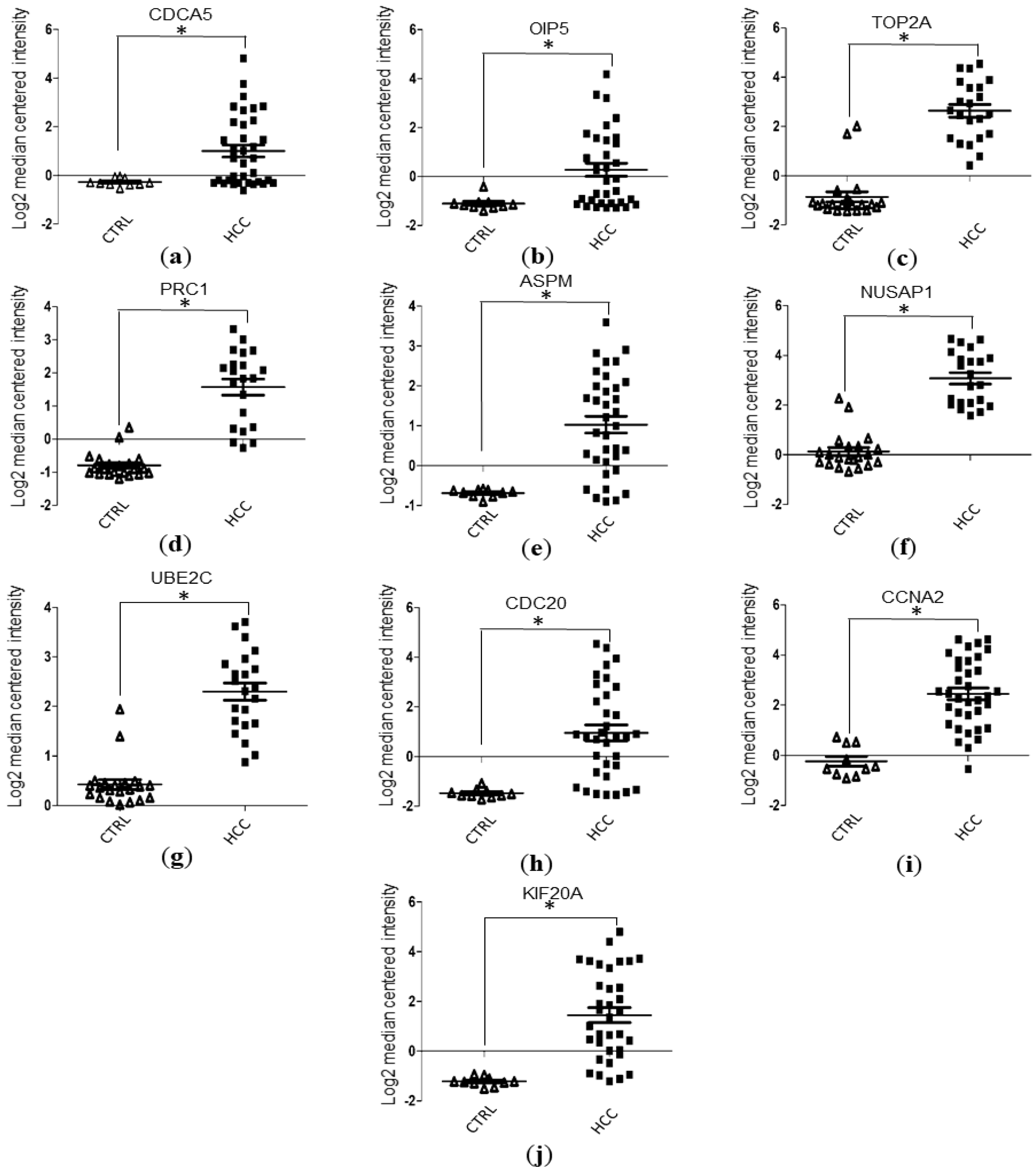
3.4. Validation of Hub Genes Expression Level with Data Mining Using Oncomine Database
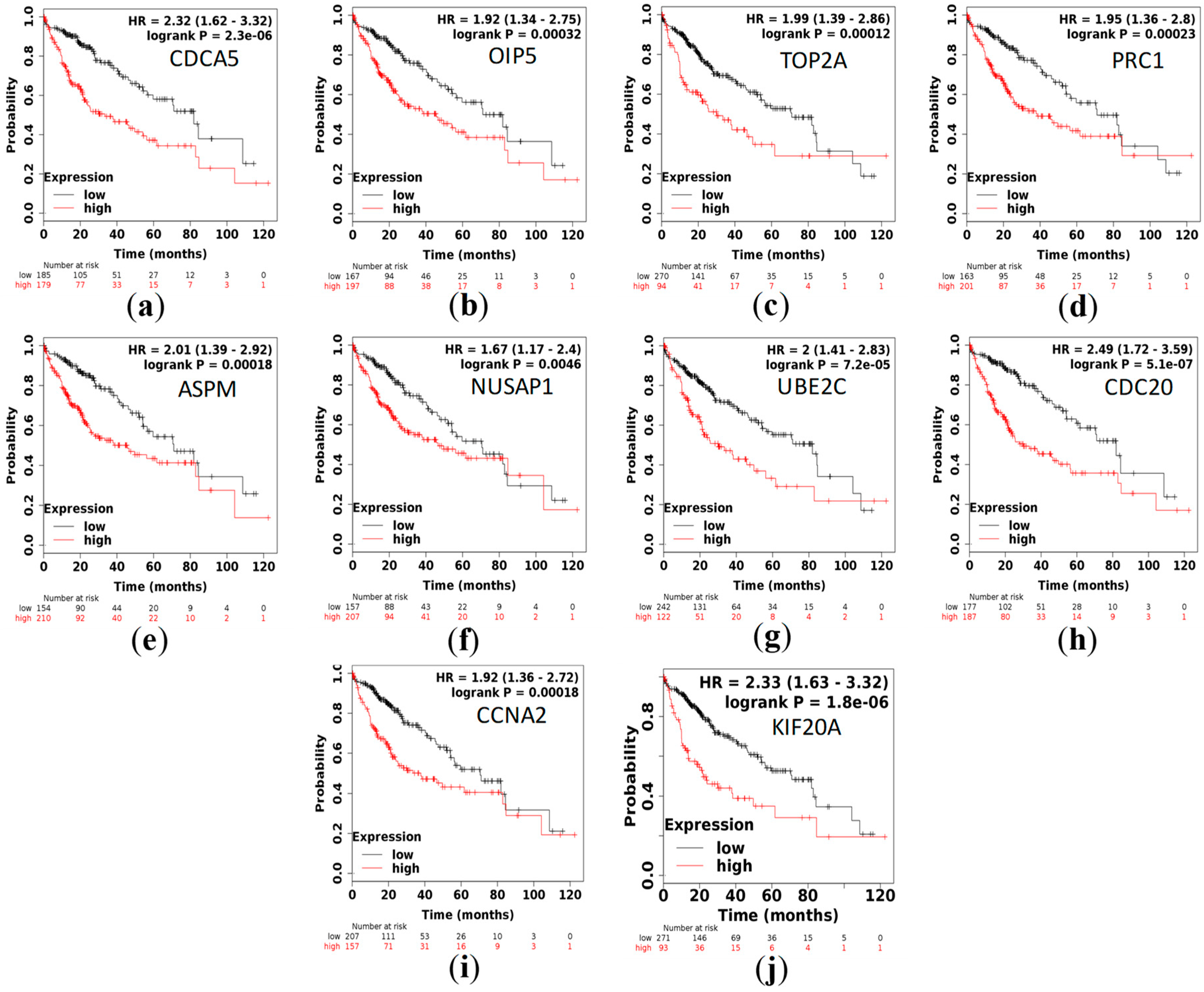
3.5. Survival Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, S.-Q.; Chen, Q.; Qin, H.-X.; Yu, Y.-Q.; Weng, J.; Mo, Q.-R.; Yin, X.-F.; Lin, Y.; Liao, W.-J. LINC00152 promotes hepatocellular carcinoma progression by regulating PI3k/Akt/mTOR signaling pathway through miR-139/PIK3CA. Am. J. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sun, V.C.; Sarna, L. Symptom management in hepatocellular carcinoma. Clin. J. Oncol. Nurs. 2008, 12, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Bertuccio, P.; Turati, F.; Carioli, G.; Rodriguez, T.; La Vecchia, C.; Malvezzi, M.; Negri, E. Global trends and predictions in hepatocellular carcinoma mortality. J. Hepatol. 2017, 67, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, V.; Gorgen, A.; Roayaie, S.; Droz Dit Busset, M.; Sapisochin, G. Liver resection and transplantation for intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 72, 364–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.R.; Chan, M.V.; Chan, C. Resection plus post-operative adjuvant transcatheter arterial chemoembolization (tace) compared with resection alone for hepatocellular carcinoma: A systematic review and meta-analysis. Cardiovasc. Intervent. Radiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.H.; Baey, S.; D’Cruz, R.T.; Shelat, V.G. Trans-arterial chemoembolization radiofrequency ablation versus surgical resection in hepatocellular carcinoma—A meta-analysis. Eur. J. Surg. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bhutiani, N.; O’Brien, S.J.; Priddy, E.E.; Egger, M.E.; Hong, Y.K.; Mercer, M.K.; McMasters, K.M.; Martin, R.C.G.; Potts, M.H.; Scoggins, C.R. Correlating serum alpha-fetoprotein in hepatocellular carcinoma with response to Yttrium-90 transarterial radioembolization with glass microspheres (TheraSphere™). HPB 2020. [Google Scholar] [CrossRef]
- Xu, Y.; Kong, Y.; Xu, J.; Li, X.; Gou, J.; Yin, T.; He, H.; Zhang, Y.; Tang, X. Doxorubicin intercalated copper diethyldithiocarbamate functionalized layered double hydroxide hybrid nanoparticles for targeted therapy of hepatocellular carcinoma. Biomater. Sci. 2019. [Google Scholar] [CrossRef]
- Lee, G.C.; Ferrone, C.R.; Vagefi, P.A.; Uppot, R.N.; Tanabe, K.K.; Lillemoe, K.D.; Blaszkowsky, L.S.; Qadan, M. Surgical resection versus ablation for early-stage hepatocellular carcinoma: A retrospective cohort analysis. Am. J. Surg. 2019. [Google Scholar] [CrossRef]
- Pan, Y.-X.; Chen, J.-C.; Fang, A.-P.; Wang, X.-H.; Chen, J.-B.; Wang, J.-C.; He, W.; Fu, Y.-Z.; Xu, L.; Chen, M.-S.; et al. A nomogram predicting the recurrence of hepatocellular carcinoma in patients after laparoscopic hepatectomy. Cancer Commun. 2019, 39, 55. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Peng, L.; Zhang, Y.; Liu, Z.; Li, W.; Chen, S.; Li, G. The identification of key genes and pathways in hepatocellular carcinoma by bioinformatics analysis of high-throughput data. Med. Oncol. 2017, 34, 101. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Kramer, J.R.; Omino, R.; Chayanupatkul, M.; Richardson, P.A.; El-Serag, H.B.; Kanwal, F. Role of age and race in the risk of hepatocellular carcinoma in veterans with hepatitis b virus infection. Clin. Gastroenterol. Hepatol. 2018, 16, 252–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, W.M.; Sellers, C.M.; Stein, S.; Lim, J.K.; Kim, H.S. Impact of direct acting antivirals on survival in patients with chronic hepatitis c and hepatocellular carcinoma. Sci. Rep. 2019, 9, 17081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Xing, H.; Wang, G.; Wang, N.; Liu, M.; Yan, C.; Li, H.; Wei, L.; Li, S.; Fan, Z.; et al. A novel online calculator based on serum biomarkers to detect hepatocellular carcinoma among patients with Hepatitis B. Clin. Chem. 2019, 65, 1543–1553. [Google Scholar] [CrossRef]
- Wang, L.; Li, H.; Zhen, Z.; Ma, X.; Yu, W.; Zeng, H.; Li, L. CXCL17 promotes cell metastasis and inhibits autophagy via the LKB1-AMPK pathway in hepatocellular carcinoma. Gene 2019, 690, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-alpha is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2018. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.F.; Li, Q.H.; Zeng, L.R.; Yang, X.Y.; Peng, P.L.; He, J.H.; Fan, B. A 4-gene prognostic signature predicting survival in hepatocellular carcinoma. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Cao, J.; Bu, Z.; Wang, Y.; Yang, H.; Jiang, J.; Li, H. Detecting prosumer-community groups in smart grids from the multiagent perspective. IEEE Trans. Syst. Man Cybern. Syst. 2019, 49, 1652–1664. [Google Scholar] [CrossRef]
- Bu, Z.; Wang, Y.; Li, H.; Jiang, J.; Wu, Z.; Cao, J. Link prediction in temporal networks: Integrating survival analysis and game theory. Inf. Sci. 2019, 498, 41–61. [Google Scholar] [CrossRef]
- Song, A.; Liu, Y.; Wu, Z.; Zhai, M.; Luo, J. A local random walk model for complex networks based on discriminative feature combinations. Expert Syst. Appl. 2019, 118, 329–339. [Google Scholar] [CrossRef]
- Hu, W.Q.; Wang, W.; Fang, D.L.; Yin, X.F. Identification of biological targets of therapeutic intervention for hepatocellular carcinoma by integrated bioinformatical analysis. Med. Sci. Monit. 2018, 24, 3450–3461. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic. Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, D.R.; Kalyana-Sundaram, S.; Mahavisno, V.; Varambally, R.; Yu, J.; Briggs, B.B.; Barrette, T.R.; Anstet, M.J.; Kincead-Beal, C.; Kulkarni, P.; et al. Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 2007, 9, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Á.; Lánczky, A.; Menyhárt, O.; Győrffy, B. Author Correction: Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 11515. [Google Scholar] [CrossRef]
- Menyhárt, O.; Nagy, Á.; Győrffy, B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 2018, 5, 181006. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Gores, G.J.; Mazzaferro, V. Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut 2014, 63, 844–855. [Google Scholar] [CrossRef]
- Lin, D.C.; Mayakonda, A.; Dinh, H.Q.; Huang, P.; Lin, L.; Liu, X.; Ding, L.W.; Wang, J.; Berman, B.P.; Song, E.W.; et al. Genomic and epigenomic heterogeneity of hepatocellular carcinoma. Cancer Res. 2017, 77, 2255–2265. [Google Scholar] [CrossRef] [Green Version]
- Baffy, G. Hepatocellular carcinoma in obesity: Finding a needle in the haystack? Adv. Exp. Med. Biol. 2018, 1061, 63–77. [Google Scholar] [CrossRef]
- Stickel, F.; Buch, S.; Nischalke, H.D.; Weiss, K.H.; Gotthardt, D.; Fischer, J.; Rosendahl, J.; Marot, A.; Elamly, M.; Casper, M.; et al. Genetic variants in PNPLA3 and TM6SF2 predispose to the development of hepatocellular carcinoma in individuals with alcohol-related cirrhosis. Am. J. Gastroenterol. 2018, 113, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-Z.; Liao, X.-W.; Wang, X.-K.; Gong, Y.-Z.; Liu, X.-G.; Yu, L.; Han, C.-Y.; Yang, C.-K.; Su, H.; Huang, K.-T.; et al. Comprehensive investigation of p53, p21, nm23, and VEGF expression in hepatitis B virus-related hepatocellular carcinoma overall survival after hepatectomy. J. Cancer 2020, 11, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Maraghy, S.A.; Adel, O.; Zayed, N.; Yosry, A.; El-Nahaas, S.M.; Gibriel, A.A. Circulatory miRNA-484, 524, 615 and 628 expression profiling in HCV mediated HCC among Egyptian patients; implications for diagnosis and staging of hepatic cirrhosis and fibrosis. J. Adv. Res. 2020, 22, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Koutb, F.; Abdel-Rahman, S.; Hassona, E.; Haggag, A. Association of c-myc and p53 gene expression and polymorphisms with hepatitis c (hcv) chronic infection, cirrhosis and hepatocellular carcinoma (hcc) stages in egypt. Asian Pac. J. Cancer Prev. 2017, 18, 2049–2057. [Google Scholar] [CrossRef]
- Fransvea, E.; Paradiso, A.; Antonaci, S.; Giannelli, G. HCC heterogeneity: Molecular pathogenesis and clinical implications. Cell. Oncol. 2009, 31, 227–233. [Google Scholar] [CrossRef]
- Jeng, K.S.; Chang, C.F.; Jeng, W.J.; Sheen, I.S.; Jeng, C.J. Heterogeneity of hepatocellular carcinoma contributes to cancer progression. Crit. Rev. Oncol./Hematol. 2015, 94, 337–347. [Google Scholar] [CrossRef]
- Cai, C.; Wang, W.; Tu, Z. Aberrantly DNA methylated-differentially expressed genes and pathways in hepatocellular carcinoma. J. Cancer 2019, 10, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Kong, J.; Qiu, Y.; Yang, X.; Wang, W.; Yan, L. Identification of core genes and outcomes in hepatocellular carcinoma by bioinformatics analysis. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Borton, M.T.; Rashid, M.S.; Dreier, M.R.; Taylor, W.R. Multiple levels of regulation of sororin by cdk1 and aurora B. J. Cell. Biochem. 2016, 117, 351–360. [Google Scholar] [CrossRef]
- Schmitz, J.; Watrin, E.; Lenart, P.; Mechtler, K.; Peters, J.M. Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr. Biol. 2007, 17, 630–636. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Wu, J.; Chagas, C.; Du, Y.; Lyu, H.; He, Y.; Qi, S.; Peng, Y.; Hu, J. CDCA5 overexpression is an Indicator of poor prognosis in patients with hepatocellular carcinoma (HCC). BMC Cancer 2018, 18, 1187. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Lee, M.J.; Yu, G.R.; Han, X.; Kim, D.G. OIP5, a target of miR-15b-5p, regulates hepatocellular carcinoma growth and metastasis through the AKT/mTORC1 and beta-catenin signaling pathways. Oncotarget 2017, 8, 18129–18144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panvichian, R.; Tantiwetrueangdet, A.; Angkathunyakul, N.; Leelaudomlipi, S. TOP2A amplification and overexpression in hepatocellular carcinoma tissues. BioMed. Res. Int. 2015, 2015, 381602. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Yeo, W.; Wong, W.L.; Wong, N.L.; Chan, K.Y.; Mo, F.K.; Koh, J.; Chan, S.L.; Chan, A.T.; Lai, P.B.; et al. TOP2A overexpression in hepatocellular carcinoma correlates with early age onset, shorter patients survival and chemoresistance. Int. J. Cancer 2009, 124, 644–652. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, F.; Xing, G.H.; Xie, P.; Zhao, N.; Yin, Y.F.; Sun, S.Y.; He, J.; Wang, Y.; Xuan, S.Y. Protein regulator of cytokinesis prc1 confers chemoresistance and predicts an unfavorable postoperative survival of hepatocellular carcinoma patients. J. Cancer 2017, 8, 801–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.Y.; Pan, H.W.; Liu, S.H.; Jeng, Y.M.; Hu, F.C.; Peng, S.Y.; Lai, P.L.; Hsu, H.C. ASPM is a novel marker for vascular invasion, early recurrence, and poor prognosis of hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 4814–4820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Yang, D.; Liu, X.; Liu, Y.; Liang, J. Expression and clinical significance of Nusap1 in hepatical carcinoma. J. Cent. South Univ. Med. Sci. 2013, 38, 876–881. [Google Scholar] [CrossRef]
- Roy, S.; Hooiveld, G.J.; Seehawer, M.; Caruso, S.; Heinzmann, F.; Schneider, A.T.; Frank, A.K.; Cardenas, D.V.; Sonntag, R.; Luedde, M.; et al. microRNA 193a-5p regulates levels of nucleolar- and spindle-associated protein 1 to suppress hepatocarcinogenesis. Gastroenterology 2018, 155, 1951–1966. [Google Scholar] [CrossRef]
- Zhuang, L.; Yang, Z. Upregulation of BUB1B, CCNB1, CDC7, CDC20, and MCM3 in tumor tissues predicted worse overall survival and disease-free survival in hepatocellular carcinoma patients. Biomed. Res. Int. 2018, 2018, 7897346. [Google Scholar] [CrossRef]
- Yang, F.; Gong, J.; Wang, G.; Chen, P.; Yang, L.; Wang, Z. Waltonitone inhibits proliferation of hepatoma cells and tumorigenesis via FXR-miR-22-CCNA2 signaling pathway. Oncotarget 2016, 7, 75165–75175. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Huang, X.; Chen, Y.; Fu, Y.; Xu, C.; Xiang, W.; Li, C.; Zhang, S.; Yu, C. Aberrant KIF20A expression might independently predict poor overall survival and recurrence-free survival of hepatocellular carcinoma. IUBMB Life 2018, 70, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hub Genes | GSE87630 | GSE89377 | GSE84598 |
|---|---|---|---|
| CDCA5 | 2.28 | 1.9 | 2.55 |
| OIP5 | 1.32 | 1.29 | 1.72 |
| TOP2A | 3.15 | 2.33 | 3.21 |
| PRC1 | 2.74 | 1.81 | 2.56 |
| ASPM | 1.53 | 1.61 | 2.25 |
| NUSAP1 | 1.82 | 1.67 | 1.53 |
| UBE2C | 1.88 | 2.4 | 2.77 |
| CDC20 | 3.01 | 2.33 | 3.09 |
| CCNA2 | 1.56 | 1.03 | 1.82 |
| KIF20A | 2.22 | 1.37 | 3.76 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Xu, J. Identification of Potentially Therapeutic Target Genes of Hepatocellular Carcinoma. Int. J. Environ. Res. Public Health 2020, 17, 1053. https://doi.org/10.3390/ijerph17031053
Li C, Xu J. Identification of Potentially Therapeutic Target Genes of Hepatocellular Carcinoma. International Journal of Environmental Research and Public Health. 2020; 17(3):1053. https://doi.org/10.3390/ijerph17031053
Chicago/Turabian StyleLi, Chengzhang, and Jiucheng Xu. 2020. "Identification of Potentially Therapeutic Target Genes of Hepatocellular Carcinoma" International Journal of Environmental Research and Public Health 17, no. 3: 1053. https://doi.org/10.3390/ijerph17031053