
Comprehensive Evaluation of Differential Methylation Analysis Methods for Bisulfite Sequencing Data



Abstract
:1. Background
2. Results and Discussion
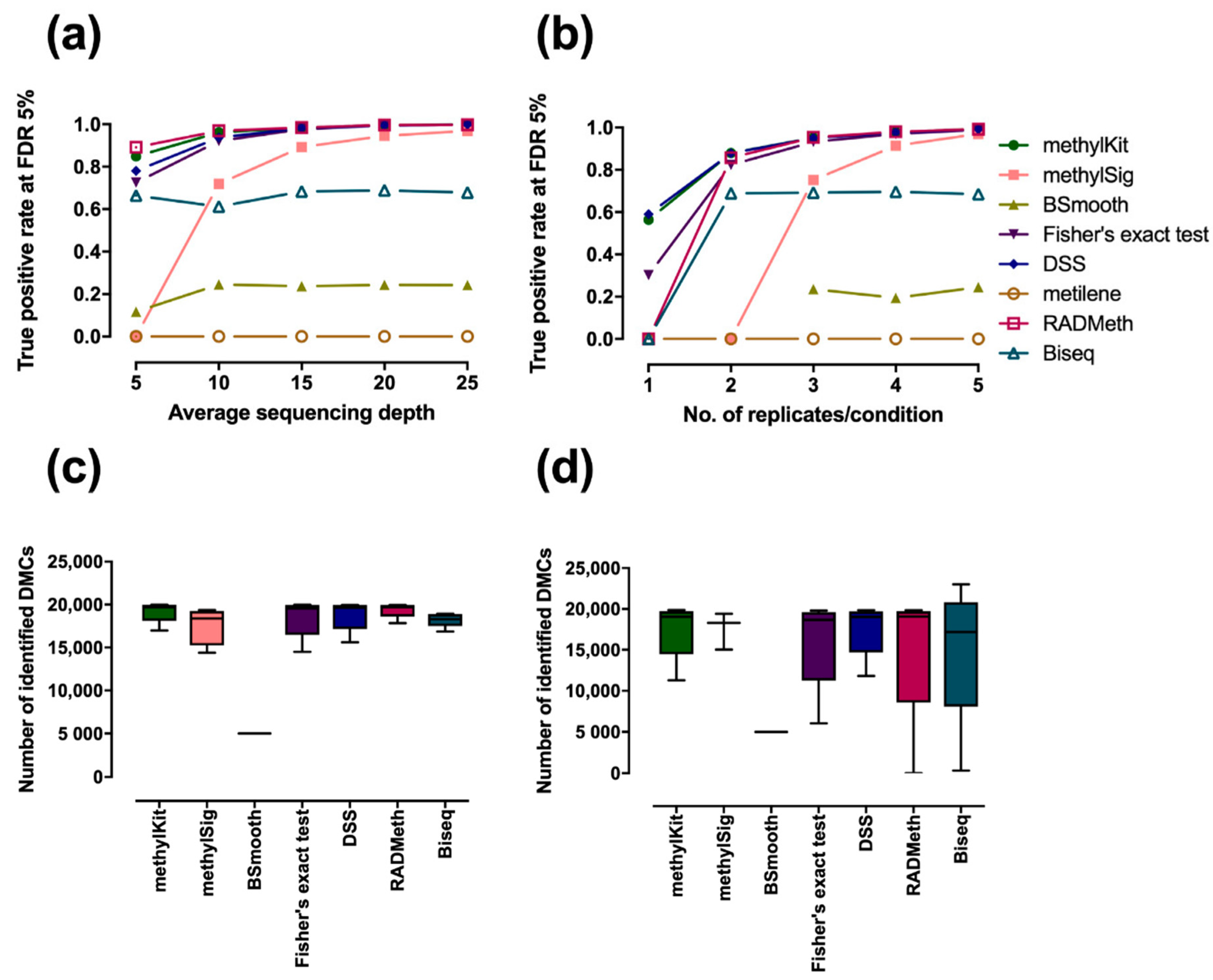
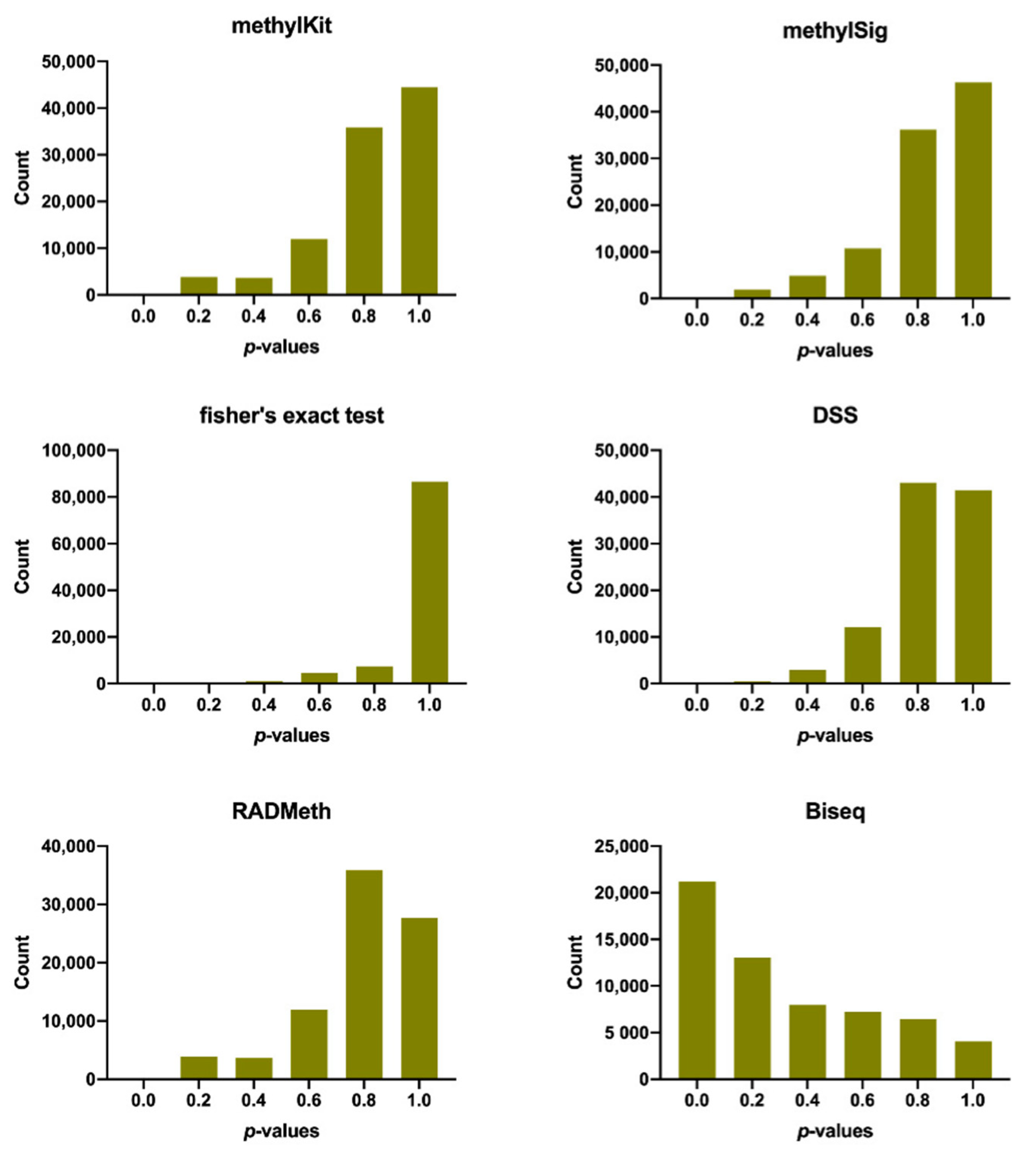
2.1. Assessment of Performance in Detecting Differentially Methylated Cytosines
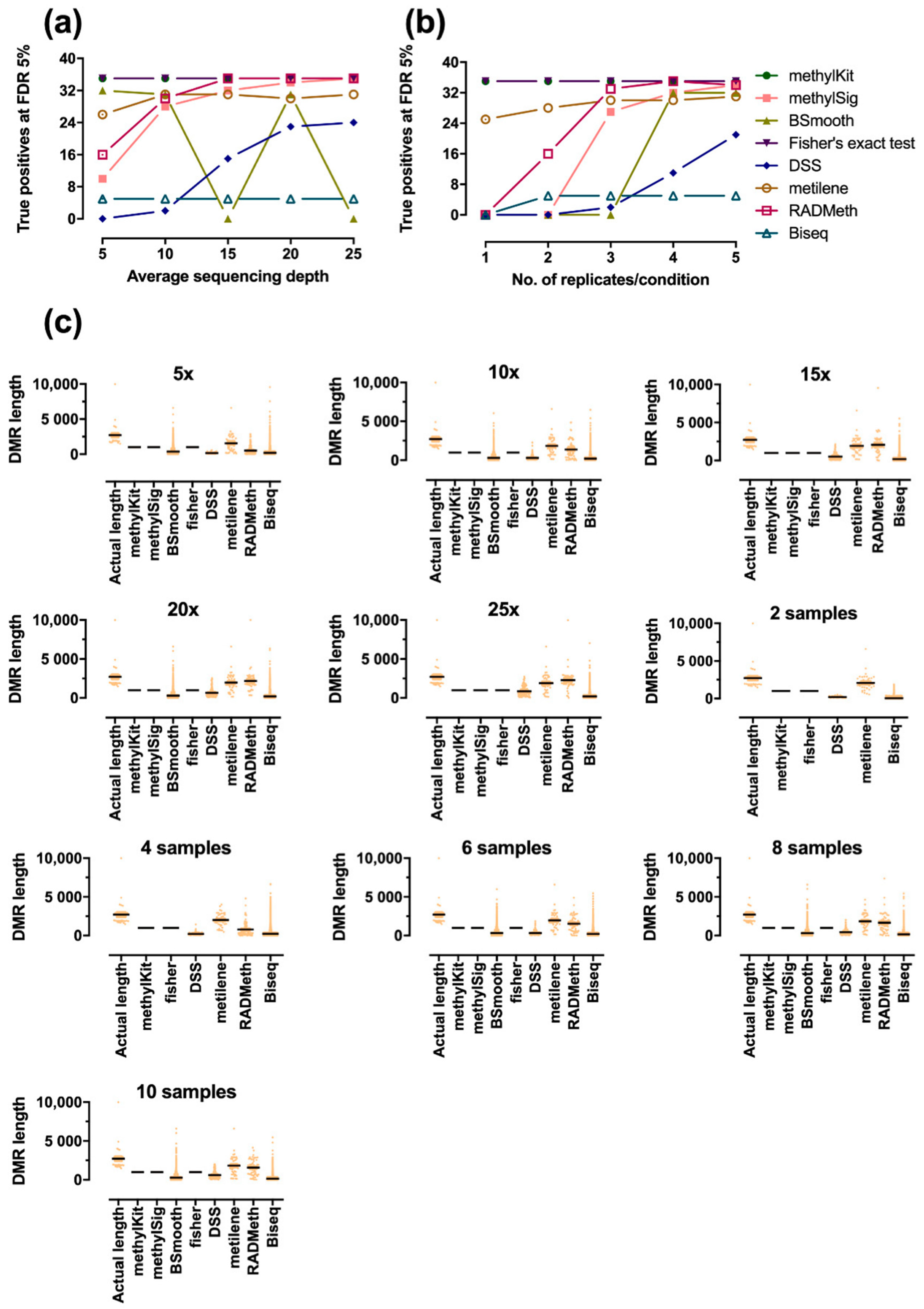
2.2. Assessment of Performance in Detecting Differentially Methylated Regions
2.3. Differential Analysis of Mouse Methylome
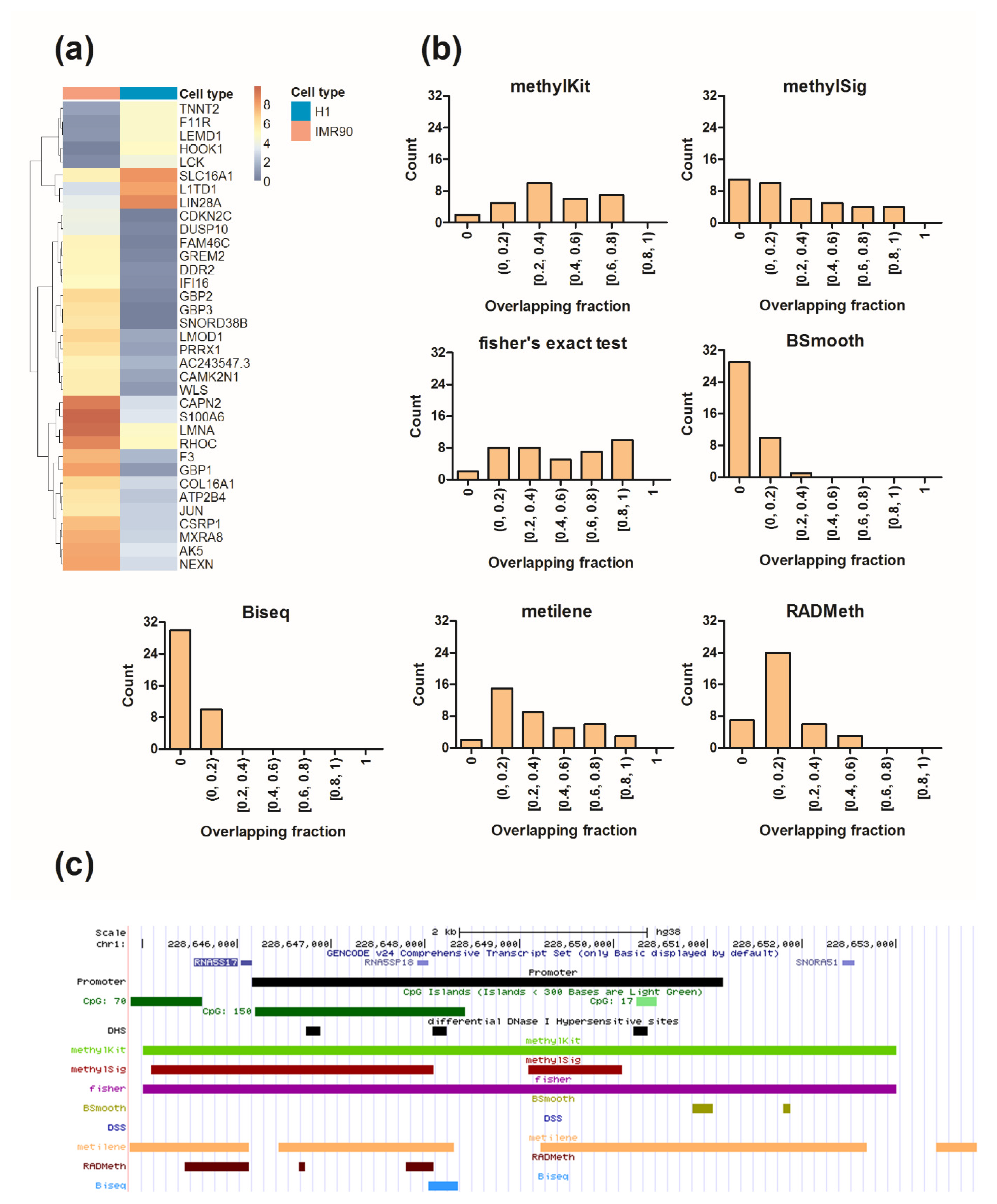
2.4. Differential Analysis of the Human Methylome
3. Conclusions
4. Materials and Methods
4.1. Simulation
4.2. RNA-Seq
4.3. DNase-Seq
4.4. BS-Seq
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krueger, F.; Kreck, B.; Franke, A.; Andrews, S.R. DNA methylome analysis using short bisulfite sequencing data. Nat. Chem. Biol. 2012, 9, 145–151. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Gabel, H.W.; Kinde, B.Z.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nat. Cell Biol. 2015, 522, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sasaki, H. Genomic imprinting in mammals: Its life cycle, molecular mechanisms and reprogramming. Cell Res. 2011, 21, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA methylation dynamics dur-ing in vivo differentiation of blood and skin stem cells. Mol. Cell. 2012, 47, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Kulis, M.; Merkel, A.; Heath, S.; Queiros, A.; Schuyler, R.P.; Castellano, G.; Beekman, R.; Raineri, E.; Esteve-Codina, A.; Clot, G.; et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat. Genet. 2015, 47, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methyla-tion prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Q.; Tennakoon, C.; Li, G.; Wong, E.; Ruan, Y.; Wei, C.L.; Sung, W.K. BatMeth: Improved mapper for bi-sulfite sequencing reads on DNA methylation. Genome Biol. 2012, 13, R82. [Google Scholar] [CrossRef] [Green Version]
- Kunde-Ramamoorthy, G.; Coarfa, C.; Laritsky, E.; Kessler, N.; Harris, R.; Xu, M.; Chen, R.; Shen, L.; Milosavljevic, A.; Waterland, R.A. Comparison and quantitative verification of mapping algorithms for whole-genome bisulfite sequencing. Nucleic Acids Res. 2014, 42, e43. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Morison, M. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic Acids Res. 2012, 40, e79. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nat. Cell Biol. 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.D.; Langmead, B.; A Irizarry, R. BSmooth: From whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012, 13, R83. [Google Scholar] [CrossRef] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Figueroa, M.E.; Rozek, L.S.; Sartor, M.A. MethylSig: A whole genome DNA methylation analysis pipeline. Bioinformatics 2014, 30, 2414–2422. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Conneely, K.N.; Wu, H. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014, 42, e69. [Google Scholar] [CrossRef] [Green Version]
- Jühling, F.; Kretzmer, H.; Bernhart, S.H.; Otto, C.; Stadler, P.F.; Hoffmann, S. metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 2016, 26, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolzhenko, E.; Smith, A.D. Using beta-binomial regression for high-precision differential methylation analysis in multifactor whole-genome bisulfite sequencing experiments. BMC Bioinform. 2014, 15, 215. [Google Scholar] [CrossRef] [Green Version]
- Hebestreit, K.; Dugas, M.; Klein, H.-U. Detection of significantly differentially methylated regions in targeted bisulfite sequencing data. Bioinformatics 2013, 29, 1647–1653. [Google Scholar] [CrossRef] [Green Version]
- Steinhauser, S.; Kurzawa, N.; Eils, R.; Herrmann, C. A comprehensive comparison of tools for differential ChIPseq analysis. Brief. Bioinform. 2016, 17, 953–966. [Google Scholar]
- Szalkowski, A.M.; Schmid, C.D. Rapid innovation in ChIPseq peakcalling algorithms is outdistancing benchmarking efforts. Brief. Bioinform. 2011, 12, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Wilbanks, E.G.; Facciotti, M.T. Evaluation of algorithm performance in ChIPseq peak detection. PLoS ONE 2010, 5, e11471. [Google Scholar] [CrossRef]
- Rapaport, F.; Khanin, R.; Liang, Y.; Pirun, M.; Krek, A.; Zumbo, P.; E Mason, C.; Socci, N.D.; Betel, D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14, R95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Delorenzi, M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinform. 2013, 14, 91. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.H.; Jhaveri, D.J.; Marshall, V.M.; Bauer, D.; Edson, J.; Narayanan, R.K.; Robinson, G.J.; Lundberg, A.E.; Bartlett, P.F.; Wray, N.; et al. A Comparative Study of Techniques for Differential Expression Analysis on RNA-Seq Data. PLoS ONE 2014, 9, e103207. [Google Scholar] [CrossRef] [Green Version]
- Dillies, M.A.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, G.; Castel, D.; Estelle, J.; et al. A compre-hensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief. Bioinform. 2013, 14, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Finotello, F.; Di Camillo, B. Measuring differential gene expression with RNA-seq: Challenges and strategies for data analysis. Brief. Funct. Genom. 2014, 14, 130–142. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Q.; Li, X.; Wang, X.; Li, J.; Zhu, X.; Sun, Z.S.; Wu, J. RRBS-Analyser: A Comprehensive Web Server for Reduced Representation Bisulfite Sequencing Data Analysis. Hum. Mutat. 2013, 34, 1606–1610. [Google Scholar] [CrossRef]
- Jiang, P.; Sun, K.; Lun, F.M.; Guo, A.M.; Wang, H.; Chan, K.A.; Chiu, R.W.; Lo, Y.D.; Sun, H. Methy-Pipe: An Integrated Bio-informatics Pipeline for Whole Genome Bisulfite Sequencing Data Analysis. PLoS ONE 2014, 9, e100360. [Google Scholar] [CrossRef]
- Saito, Y.; Tsuji, J.; Mituyama, T. Bisulfighter: Accurate detection of methylated cytosines and dif-ferentially methylated regions. Nucleic Acids Res. 2014, 42, e45. [Google Scholar] [CrossRef]
- Chen, Y.; Negre, N.; Li, Q.; Mieczkowska, J.O.; Slattery, M.; Liu, T.; Zhang, Y.; Kim, T.K.; He, H.H.; Zieba, J.; et al. Systematic evaluation of fac-tors influencing ChIP-seq fidelity. Nat. Methods 2012, 9, 609–614. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful ap-proach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.L.; Ren, B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Xi, Y.; Rodriguez, B.; Park, H.J.; Tong, P.; Meong, M.; Goodell, M.A.; Li, W. MOABS: Model based analysis of bisulfite sequencing data. Genome Biol. 2014, 15, R38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neu-rons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992. [Google Scholar] [CrossRef] [Green Version]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Reversing DNA Methylation: Mechanisms, Genomics, and Biological Functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- ENCODE. Available online: https://www.encodeproject.org (accessed on 3 April 2019).
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nat. Cell Biol. 2012, 489, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Piao, Y.; Lee, S.K.; Lee, E.J.; Robertson, K.D.; Shi, H.; Ryu, K.H.; Choi, J.H. CAME: Identification of chroma-tin accessibility from nucleosome occupancy and methylome sequencing. Bioinformatics 2017, 33, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates in-trinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015, 43, W589–W598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://neomorph.salk.edu/human_methylome/data.html (accessed on 4 January 2020).
- Available online: https://sourceforge.net/projects/dmrs/ (accessed on 4 January 2020).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Version | Model Assumption | Differential Methylation Test | Segmentation | Language | Smoothing |
|---|---|---|---|---|---|---|
| Fisher’s | 1.8.2 | - | Fisher’s exact test | tilling window | R | No |
| BSmooth | 1.8.2 | binomial distribution | modified t-test | merging consecutive CpGs | R | Yes |
| methylKit | 0.99.2 | logistic regression | logistic regression test | tilling window or predefined regions | R | No |
| methylSig | 0.4.4 | beta-binomial model | likelihood ratio test | tilling window | R | No |
| DSS | 2.12.0 | Bayesian hierarchical model | Wald test | merging CpGs based on p-value | R | No |
| metilene | 0.2–6 | Nonparametric method | 2D Kolmogorov–Smirnov | circular binary segmentation | C | No |
| RADMeth | - | beta-binomial regression | log-likelihood ratio test | correlation between p-value pairs within a bin | C++ | No |
| Biseq | 1.12.0 | Beta regression model | Wald test | merging consecutive CpGs | R | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piao, Y.; Xu, W.; Park, K.H.; Ryu, K.H.; Xiang, R. Comprehensive Evaluation of Differential Methylation Analysis Methods for Bisulfite Sequencing Data. Int. J. Environ. Res. Public Health 2021, 18, 7975. https://doi.org/10.3390/ijerph18157975
Piao Y, Xu W, Park KH, Ryu KH, Xiang R. Comprehensive Evaluation of Differential Methylation Analysis Methods for Bisulfite Sequencing Data. International Journal of Environmental Research and Public Health. 2021; 18(15):7975. https://doi.org/10.3390/ijerph18157975
Chicago/Turabian StylePiao, Yongjun, Wanxue Xu, Kwang Ho Park, Keun Ho Ryu, and Rong Xiang. 2021. "Comprehensive Evaluation of Differential Methylation Analysis Methods for Bisulfite Sequencing Data" International Journal of Environmental Research and Public Health 18, no. 15: 7975. https://doi.org/10.3390/ijerph18157975
APA StylePiao, Y., Xu, W., Park, K. H., Ryu, K. H., & Xiang, R. (2021). Comprehensive Evaluation of Differential Methylation Analysis Methods for Bisulfite Sequencing Data. International Journal of Environmental Research and Public Health, 18(15), 7975. https://doi.org/10.3390/ijerph18157975