
Determination of Novel Anti-Cancer Agents by Targeting OGG1 Enzyme Using Integrated Bioinformatics Methods

 , , and
, , and
Abstract
:1. Introduction
2. Results
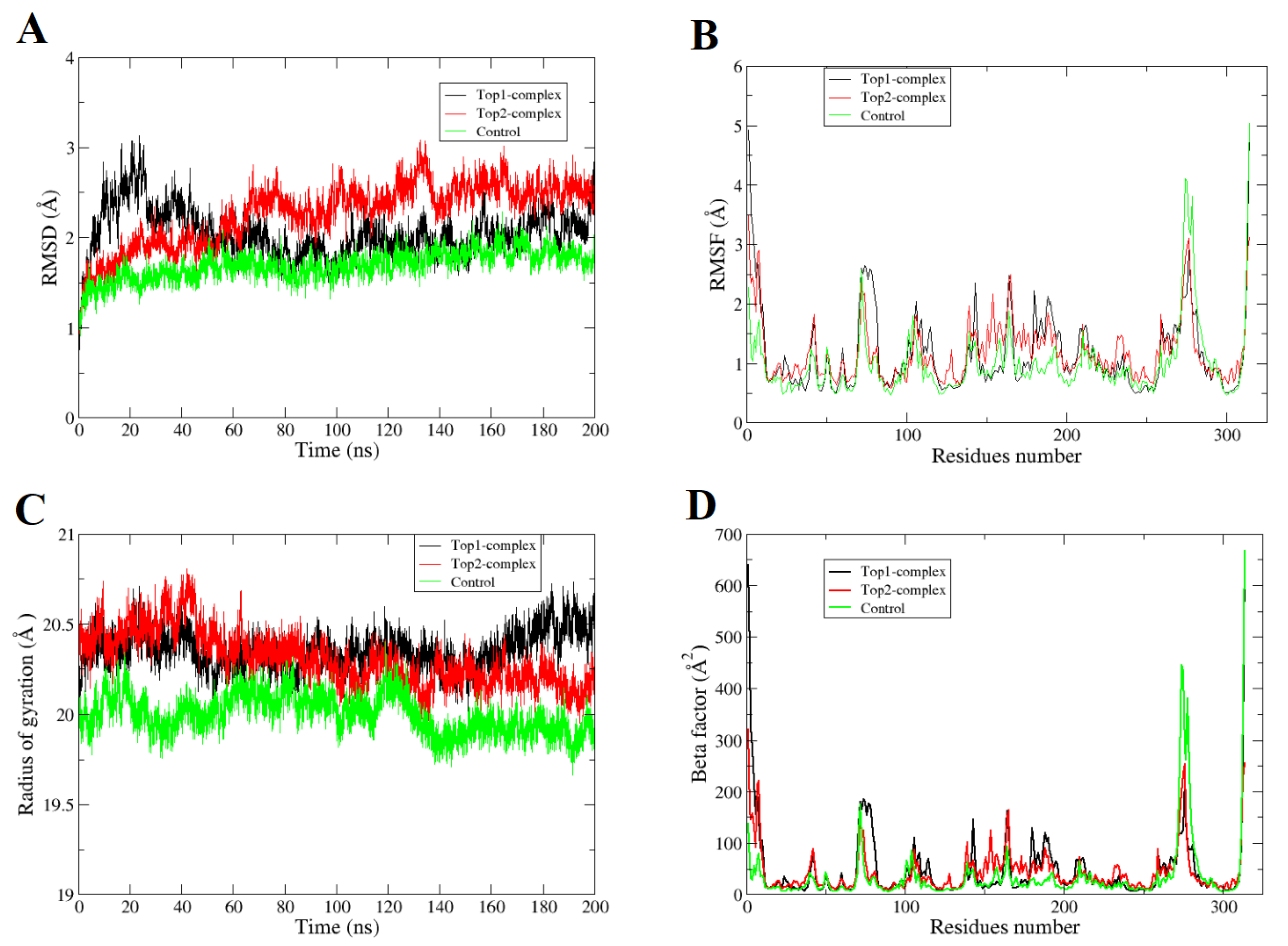
2.1. Molecular Dynamics Simulations
2.2. Hydrogen Bonds Analysis
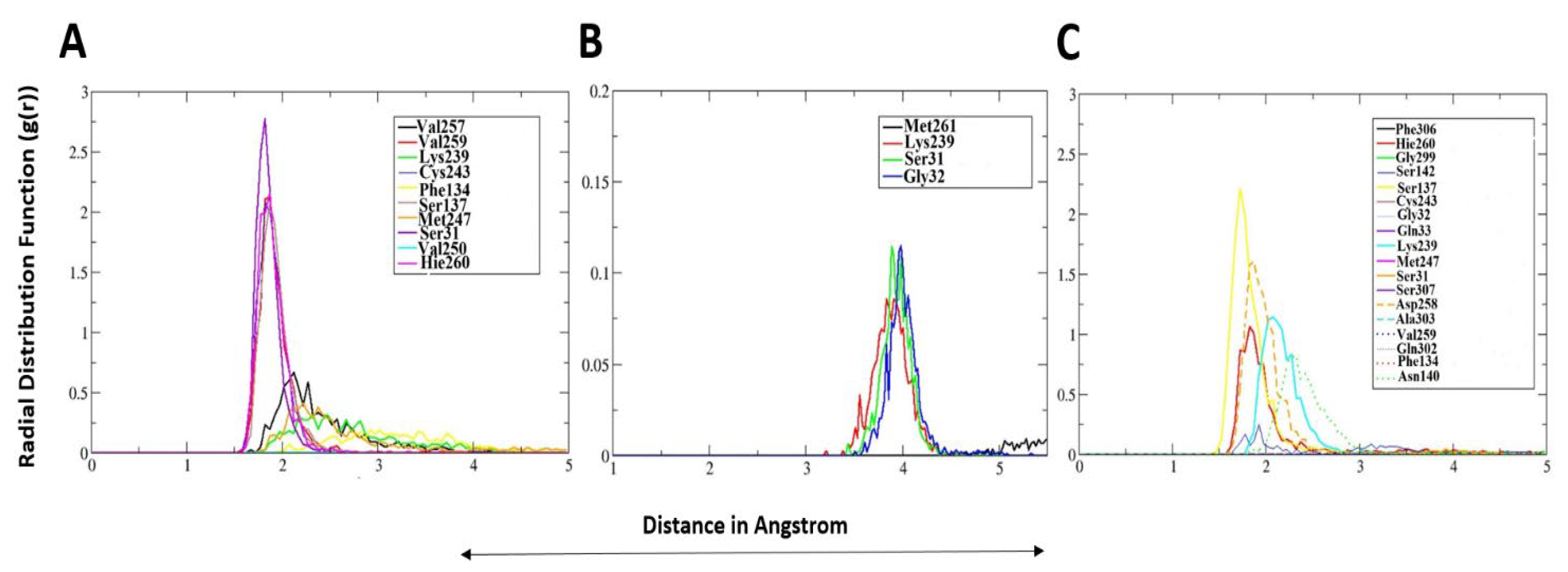
2.3. Radial Distribution Function-g(r)
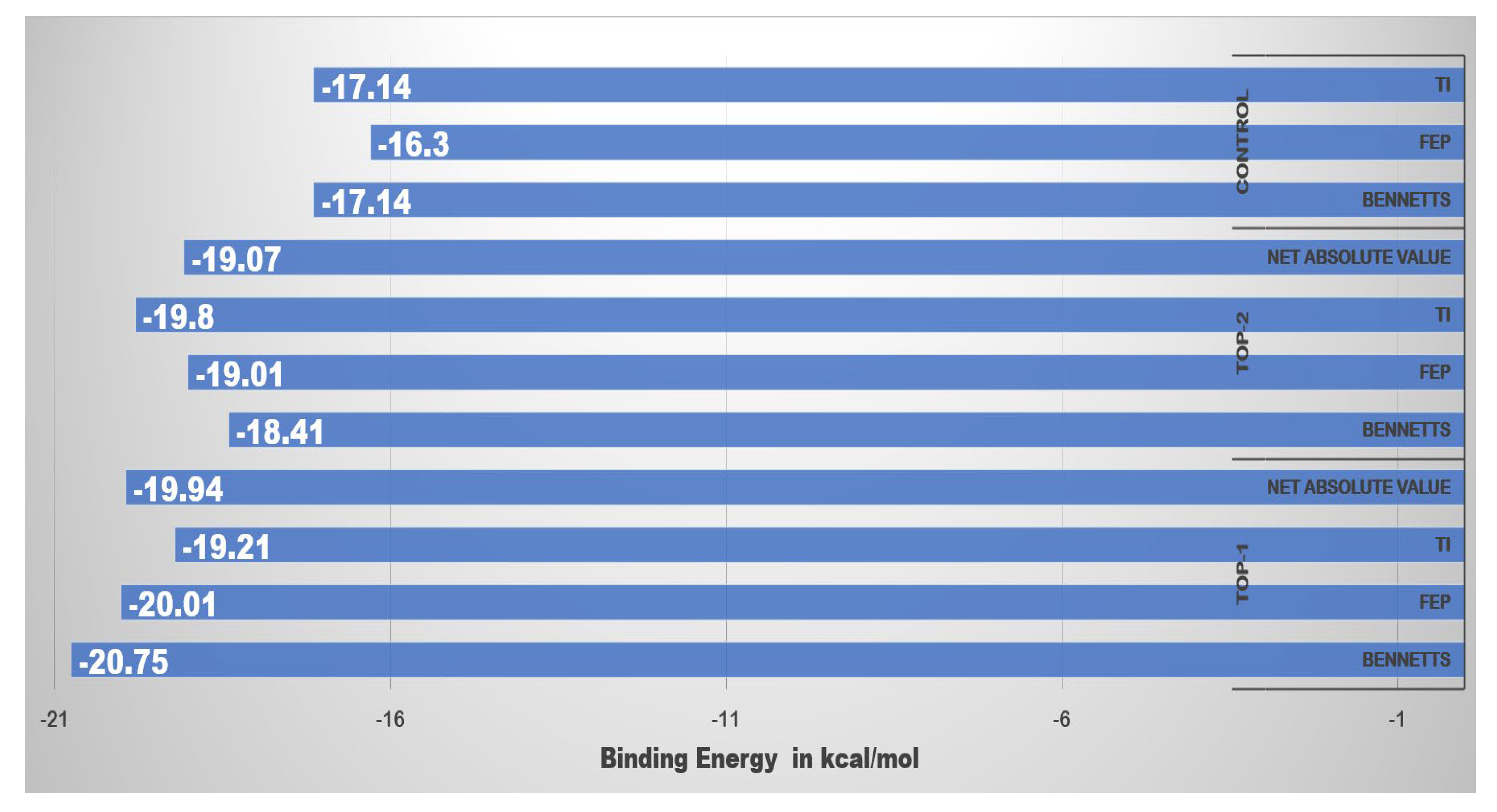
2.4. MM/GBSA and MM/PBSA Binding Free Energies
2.5. Decomposition of MM/GBSA Binding Energy
2.6. WaterSwap Binding Energies
2.7. In Silico Site Directed Mutagenesis
2.8. Computational Pharmacokinetics Studies
3. Materials and Methods
3.1. OGG1 Structure Retrieval and Preparation for Virtual Screening
3.2. Inhibitors Library Preparation
3.3. Molecular Docking for Inhibition Studies
3.4. Molecular Dynamics Simulation Study
3.5. Estimating MM/GBSA and MM/PBSA Binding Free Energies
3.6. WaterSwap Binding Energy Predictions
3.7. In Silico Site-directed Mutagenesis
3.8. Predictions about Compounds Pharmacokinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive oxygen species: A key constituent in cancer survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef]
- Steenken, S.; Jovanovic, S.V. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Damia, G.; D’Incalci, M. Targeting DNA repair as a promising approach in cancer therapy. Eur. J. Cancer 2007, 43, 1791–1801. [Google Scholar] [CrossRef]
- Murai, J.; Shar-yin, N.H.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Mansour, W.Y.; Rhein, T.; Dahm-Daphi, J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010, 38, 6065–6077. [Google Scholar] [CrossRef]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visnes, T.; Benítez-Buelga, C.; Cázares-Körner, A.; Sanjiv, K.; Hanna, B.M.F.; Mortusewicz, O.; Rajagopal, V.; Albers, J.J.; Hagey, D.W.; Bekkhus, T.; et al. Targeting OGG1 arrests cancer cell proliferation by inducing replication stress. Nucleic Acids Res. 2020, 48, 12234–12251. [Google Scholar] [CrossRef]
- Hanna, B.M.F.; Helleday, T.; Mortusewicz, O. OGG1 Inhibitor TH5487 Alters OGG1 Chromatin Dynamics and Prevents Incisions. Biomolecules 2020, 10, 1483. [Google Scholar] [CrossRef]
- Yu, W.; MacKerell, A.D. Computer-aided drug design methods. In Antibiotics; Springer: Manhattan, NY, USA, 2017; pp. 85–106. [Google Scholar]
- Suleman, M.; ul Qamar, M.T.; Shoaib Saleem, S.A.; Ali, S.S.; Khan, H.; Akbar, F.; Khan, W.; Alblihy, A.; Alrumaihi, F.; Waseem, M. Mutational Landscape of Pirin and Elucidation of the Impact of Most Detrimental Missense Variants That Accelerate the Breast Cancer Pathways: A Computational Modelling Study. Front. Mol. Biosci. 2021, 8, 692835. [Google Scholar] [CrossRef]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; ul Qamar, M.T. Pan-Vaccinomics Approach Towards a Universal Vaccine Candidate Against WHO Priority Pathogens to Address Growing Global Antibiotic Resistance. Comput. Biol. Med. 2021, 136, 104705. [Google Scholar] [CrossRef]
- Alamri, M.A.; ul Qamar, M.T.; Afzal, O.; Alabbas, A.B.; Riadi, Y.; Alqahtani, S.M. Discovery of anti-MERS-CoV small covalent inhibitors through pharmacophore modeling, covalent docking and molecular dynamics simulation. J. Mol. Liq. 2021, 330, 115699. [Google Scholar] [CrossRef]
- Mumtaz, A.; Ashfaq, U.A.; Qamar, M.T.; Anwar, F.; Gulzar, F.; Amjad, M.A.; Saari, N.; Pervez, M.T. MPD3: A useful medicinal plants database for drug designing. Nat. Prod. Res. 2020, 34, 1051. [Google Scholar] [CrossRef]
- RK, M.; AK, N. In silico evaluation of multispecies toxicity of natural compounds. Drug Chem. Toxicol. 2019, 21, 1–7. [Google Scholar]
- Khan, W.; Ashfaq, U.A.; Aslam, S.; Saif, S.; Aslam, T.; Tusleem, K.; Maryam, A.; ul Qamar, M.T. Anticancer screening of medicinal plant phytochemicals against Cyclin-Dependent Kinase-2 (CDK2): An in-silico approach. Adv. Life Sci. 2017, 4, 113–119. [Google Scholar]
- Riaz, M.; Ashfaq, U.A.; Qasim, M.; Yasmeen, E.; Ul Qamar, M.T.; Anwar, F. Screening of medicinal plant phytochemicals as natural antagonists of p53-MDM2 interaction to reactivate p53 functioning. Anticancer. Drugs 2017, 28, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. In Molecular Modeling of Proteins; Springer: Manhattan, NY, USA, 2008; pp. 365–382. [Google Scholar]
- Khalid, R.R.; ul Qamar, M.T.; Maryam, A.; Ashique, A.; Anwar, F.; Geesi, M.H.; Siddiqi, A. Comparative Studies of the Dynamics Effects of BAY60-2770 and BAY58-2667 Binding with Human and Bacterial H-NOX Domains. Molecules 2018, 23, 2141. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muneer, I.; Ul Qamar, M.T.; Tusleem, K.; Abdul Rauf, S.; Hussain, H.M.J.; Siddiqi, A.R. Discovery of selective inhibitors for cyclic AMP response element-binding protein: A combined ligand and structure-based resources pipeline. Anticancer. Drugs 2019, 30, 363–373. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Tahir ul Qamar, M.; Salmas, R.E.; Tariq, Q.; Anwar, F.; Ashfaq, U.A. Investigating the molecular mechanism of staphylococcal DNA gyrase inhibitors: A combined ligand-based and structure-based resources pipeline. J. Mol. Graph. Model. 2018, 85, 122–129. [Google Scholar] [CrossRef]
- Woods, C.J.; Malaisree, M.; Michel, J.; Long, B.; McIntosh-Smith, S.; Mulholland, A.J. Rapid decomposition and visualisation of protein-ligand binding free energies by residue and by water. Faraday Discuss. 2014, 169, 477–499. [Google Scholar] [CrossRef] [Green Version]
- Woods, C.J.; Malaisree, M.; Hannongbua, S.; Mulholland, A.J. A water-swap reaction coordinate for the calculation of absolute protein-ligand binding free energies. J. Chem. Phys. 2011, 134, 02B611. [Google Scholar] [CrossRef] [Green Version]
- Khan, R.J.; Jha, R.K.; Amera, G.; Jain, M.; Singh, E.; Pathak, A.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. Targeting SARS-Cov-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like Proteinase and 2’-O-RiboseMethyltransferase. J. Biomol. Struct. Dyn. 2020, 39, 2679–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiorov, V.N.; Crippen, G.M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Pant, S.; Singh, M.; Ravichandiran, V.; Murty, U.S.N.; Srivastava, H.K. Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn. 2020, 39, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O. V Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Iqbal, S.; Shamim, A.; Azam, S.S.; Wadood, A. Identification of potent inhibitors for chromodomain-helicase- DNA-binding protein 1-like through moleculardocking studies. Med. Chem. Res. 2016, 25, 2924–2939. [Google Scholar] [CrossRef]
- Wade, R.C.; Goodford, P.J. The role of hydrogen-bonds in drug binding. Prog. Clin. Biol. Res. 1989, 289, 433–444. [Google Scholar] [PubMed]
- Abbasi, S.; Raza, S.; Azam, S.S.; Liedl, K.R.; Fuchs, J.E. Interaction mechanisms of a melatonergic inhibitor in the melatonin synthesis pathway. J. Mol. Liq. 2016, 221, 507–517. [Google Scholar] [CrossRef]
- Raza, S.; Azam, S.S. AFD: An application for bi-molecular interaction using axial frequency distribution. J. Mol. Model. 2018, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM_PBSA and MM_GBSA Methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Modeling 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Joshi, R.S.; Jagdale, S.S.; Bansode, S.B.; Shankar, S.S.; Tellis, M.B.; Pandya, V.K.; Chugh, A.; Giri, A.P.; Kulkarni, M.J. Discovery of Potential Multi-Target-Directed Ligands by Targeting Host-specific SARS-CoV-2 Structurally Conserved Main Protease$. J. Biomol. Struct. Dyn. 2021, 39, 3099–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abro, A.; Azam, S.S. Binding free energy based analysis of arsenic (+ 3 oxidation state) methyltransferase with S-adenosylmethionine. J. Mol. Liq. 2016, 220, 375–382. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Jia, C.-Y.; Li, J.-Y.; Hao, G.-F.; Yang, G.-F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 2019, 25, 248–258. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Whitty, A. Growing PAINS in academic drug discovery. Future Med. Chem. 2011, 3, 797–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology; Springer: Manhattan, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Mumtaz, A.; Ashfaq, U.A.; ul Qamar, M.T.; Anwar, F.; Gulzar, F.; Ali, M.A.; Saari, N.; Pervez, M.T. MPD3: A useful medicinal plants database for drug designing. Nat. Prod. Res. 2017, 31, 1228–1236. [Google Scholar] [CrossRef]
- Lyu, C.; Chen, T.; Qiang, B.; Liu, N.; Wang, H.; Zhang, L.; Liu, Z. CMNPD: A comprehensive marine natural products database towards facilitating drug discovery from the ocean. Nucleic Acids Res. 2021, 49, D509–D515. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Visualizer; BIOVIA Discovery Studio: San Diego, CA, USA, 2017. [Google Scholar]
- Spławiński, J.; Kuźniar, J.; Filipiak, K.; Zieliński, W. Evaluation of drug toxicity in clinical trials. Sci. Eng. Ethics 2006, 12, 139–145. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Giambasu, G.; et al. d 2020. 2020. Available online: https://ambermd.org/doc12/Amber20.pdf (accessed on 8 December 2021).
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Kouetcha, D.N.; Ramézani, H.; Cohaut, N. Ultrafast scalable parallel algorithm for the radial distribution function histogramming using MPI maps. J. Supercomput. 2017, 73, 1629–1653. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z.H. Interaction entropy: A new paradigm for highly efficient and reliable computation of protein--ligand binding free energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef]
- Kiani, Y.S.; Ranaghan, K.E.; Jabeen, I.; Mulholland, A.J. Molecular Dynamics Simulation Framework to Probe the Binding Hypothesis of CYP3A4 Inhibitors. Int. J. Mol. Sci. 2019, 20, 4468. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | GOLD Score | Autodock Vina Binding Free Energy (kcal/mol) | ASP | CHEMPLP | Chem Score |
|---|---|---|---|---|---|
 Top-1 | 79 | −11.6 | 18.71 | 56.88 | 17.84 |
 Top-2 | 76 | −10.7 | 16.10 | 54.89 | 16.20 |
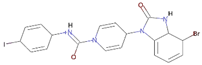 Control | 72 | −9.0 | 16.30 | 51.9 | 14.84 |
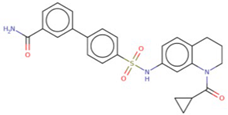 | 74 SU0268 | −9.8 | 17.37 | 50.74 | 15.87 |
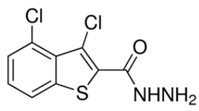 | O8 | −11.54 | 19.57 | 53.97 | 17.52 |
| MM/GBSA | |||||||
|---|---|---|---|---|---|---|---|
| Compound | ΔG Binding (kcal/mol) | ΔG Electrostatic (kcal/mol) | ΔG Bind van der Waals (kcal/mol) | ΔG Gas Phase (kcal/mol) | ΔG Polar Solvation (kcal/mol) | ΔG Non-Polar Solvation (kcal/mol) | ΔG Solvation (kcal/mol) |
| Top-1 | −28.10 | −25.26 | −41.42 | −66.69 | 44.40 | −5.80 | 38.59 |
| Top-2 | −50.14 | −17.00 | −60.47 | −77.47 | 34.54 | −7.21 | 27.32 |
| Control | −46.91 | −23.10 | −56.70 | −79.81 | 38.54 | −5.64 | 32.90 |
| MM/PBSA | |||||||
| Top-1 | −23.38 | −25.26 | −41.42 | −66.69 | 47.85 | −4.54 | 43.30 |
| Top-2 | −35.29 | −17.00 | −60.47 | −77.47 | 47.50 | −5.32 | 42.17 |
| Control | −38.20 | −23.10 | −56.70 | −79.81 | 45.55 | −3.94 | 41.60 |
| Ligand/Residue | Top-1 | Alanine Scanning Results | Top-2 | Alanine Scanning Results |
|---|---|---|---|---|
| Ligand | −15.06 | NA | −24.28 | NA |
| Ile142 | −3.209 | −2.14 | −4.38 | −2.80 |
| Phe134 | −1.90 | −1.20 | −2.74 | −1.11 |
| Phe306 | −1.77 | −0.98 | −2.84 | −1.44 |
| Ala143 | −1.19 | −1.0 | −1.75 | −1.12 |
| Cys243 | −0.97 | NA | −1.55 | NA |
| Gln33 | −0.82 | NA | −1.50 | NA |
| Ile145 | −0.80 | NA | −1.35 | NA |
| Met247 | −0.71 | NA | −1.28 | NA |
| His260 | −0.47 | 1.0 | −1.10 | −1.00 |
| Leu122 | −0.45 | NA | −0.87 | NA |
| Ala303 | −0.41 | NA | −0.80 | NA |
| Leu246 | −0.39 | NA | −0.73 | NA |
| Gly32 | −0.35 | −1.04 | −0.70 | −1.10 |
| Pro256 | −0.33 | −1.0 | −0.61 | −1.01 |
| Leu310 | −0.25 | NA | −0.55 | NA |
| Phe35 | −0.22 | 0.12 | −0.54 | −0.41 |
| Val240 | −0.21 | NA | −0.50 | NA |
| Ser31 | −0.21 | 0.11 | −0.48 | 1.18 |
| Property | Compounds | ||
|---|---|---|---|
| Physicochemical Properties | Top-1 | Top-2 | Control |
| Formula | C22H19N3O2S3 | C27H28BrN5O3S | C19H19BrIN4O2 |
| Molecular weight | 453.60 g/mol | 582.51 g/mol | 542.19 g/mol |
| Num. heavy atoms | 30 | 37 | 27 |
| Num. arom. heavy atoms | 22 | 22 | 11 |
| Fraction Csp3 | 0.14 | 0.22 | 0.32 |
| Num. rotatable bonds | 8 | 11 | 4 |
| Num. H-bond acceptors | 3 | 6 | 2 |
| Num. H-bond donors | 1 | 2 | 2 |
| Molar Refractivity | 123.71 | 149.71 | 118.21 |
| TPSA | 164.21 Å2 | 112.67 Å2 | 70.77 Å2 |
| Lipophilicity | |||
| Consensus Log Po/w | 4.22 | 5.00 | 2.38 |
| Water Solubility | Moderately soluble | Moderately soluble | Moderately soluble |
| Pharmacokinetics | |||
| GI absorption | High | High | High |
| BBB permeant | No | No | Yes |
| P-gp substrate | No | No | Yes |
| CYP1A2 inhibitor | No | No | No |
| CYP2C19 inhibitor | Yes | Yes | No |
| CYP2C9 inhibitor | Yes | Yes | No |
| CYP2D6 inhibitor | No | No | No |
| CYP3A4 inhibitor | No | No | No |
| Log Kp (skin permeation) | −4.93 cm/s | −4.80 cm/s | −7.34 cm/s |
| Druglikensess | |||
| Lipinski | Yes; 0 violation | Yes; 1 violation: MW > 500 | Yes; 1 violation: MW > 500 |
| Medicinal Chemistry | |||
| PAINS | 0 alert | 0 alert | 1 alert acyl_het_A |
| Synthetic accessibility | 3.86 | 3.72 | 4.76 |
| Toxicity | |||
| Hepatotoxicity | No | Yes | Yes |
| Skin sensitisation | No | No | No |
| T. pyriformis toxicity | 0.292 log ug/L | 0.285 log ug/L | 0.315 log ug/L |
| Ames toxicity | No | No | No |
| Minnow toxicity | 0.69 log mM | −1.164 log mM | −1.14 log mM |
| Carcino mouse | No | No | No |
| Excretion | |||
| Total clearance | 0.763 log mL/min/kg | 0.026 log mL/min/kg | 0.031 log mL/min/kg |
| Renal OCT2 substrate | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhseen, Z.T.; Ali, M.H.; Jaber, N.R.; Mashrea, D.S.; Alfalki, A.M.; Li, G. Determination of Novel Anti-Cancer Agents by Targeting OGG1 Enzyme Using Integrated Bioinformatics Methods. Int. J. Environ. Res. Public Health 2021, 18, 13290. https://doi.org/10.3390/ijerph182413290
Muhseen ZT, Ali MH, Jaber NR, Mashrea DS, Alfalki AM, Li G. Determination of Novel Anti-Cancer Agents by Targeting OGG1 Enzyme Using Integrated Bioinformatics Methods. International Journal of Environmental Research and Public Health. 2021; 18(24):13290. https://doi.org/10.3390/ijerph182413290
Chicago/Turabian StyleMuhseen, Ziyad Tariq, Mustafa Hussein Ali, Nawar Rushdi Jaber, Dheyaa Shakir Mashrea, Ali Mamoon Alfalki, and Guanglin Li. 2021. "Determination of Novel Anti-Cancer Agents by Targeting OGG1 Enzyme Using Integrated Bioinformatics Methods" International Journal of Environmental Research and Public Health 18, no. 24: 13290. https://doi.org/10.3390/ijerph182413290