
Analysis of Single Nucleotide Variants (SNVs) Induced by Exposure to PM10 in Lung Epithelial Cells Using Whole Genome Sequencing


 ,
,  , ,
, , 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Method
2.1. Cell Culture and PM10 Treatment
2.2. Next Generation Sequencing (NGS) Analysis Workflow
2.2.1. Library Construction
2.2.2. Clustering and Sequencing
2.2.3. Generation of Raw Data
2.2.4. Read Mapping
2.2.5. SNVs, Small Indels Calling and Annotation
2.2.6. Structural Variant Calling
2.3. Data Analysis
3. Results
3.1. Overview of Whole Genome Sequencing Results
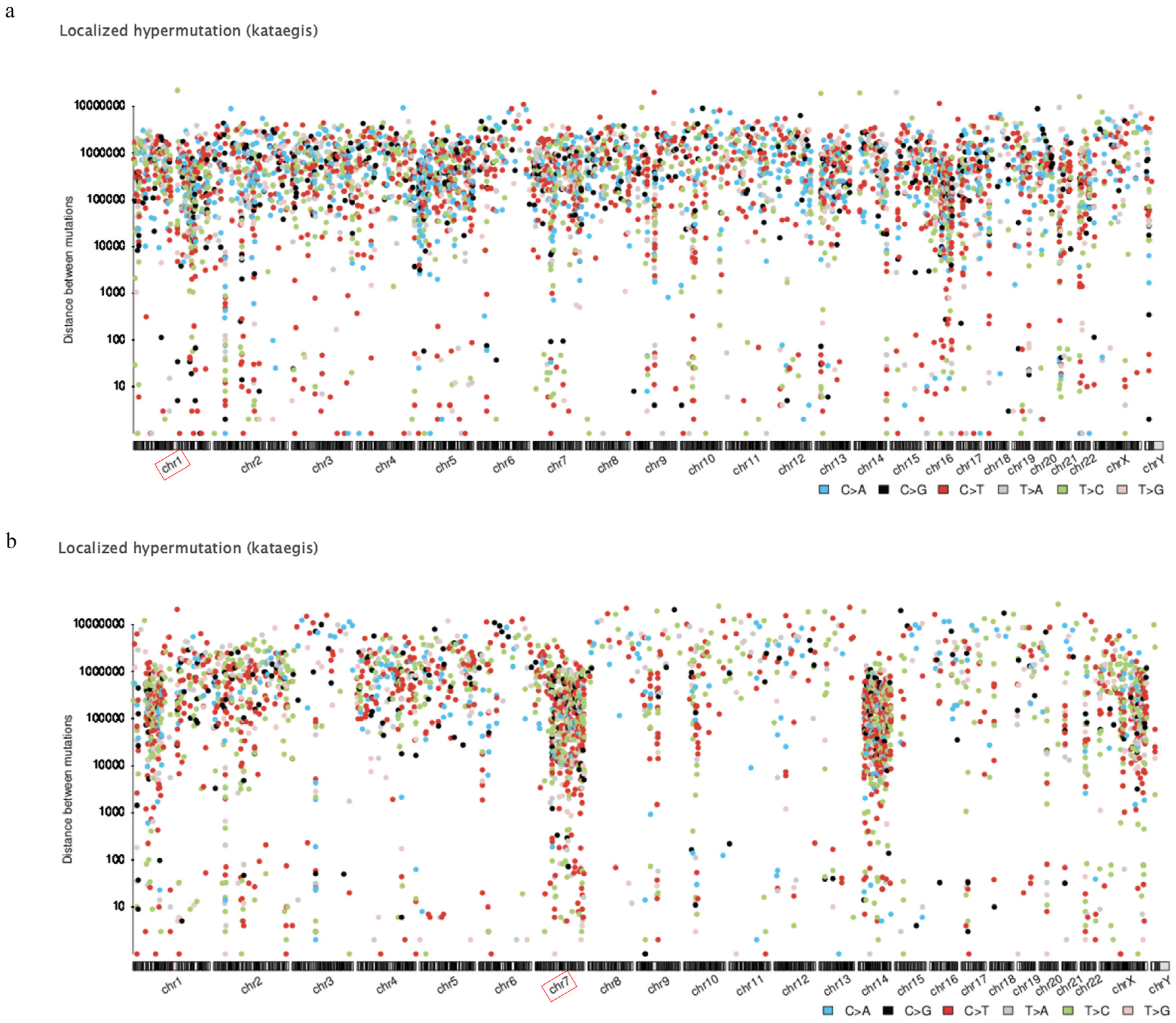
3.2. Localized Hypermutation
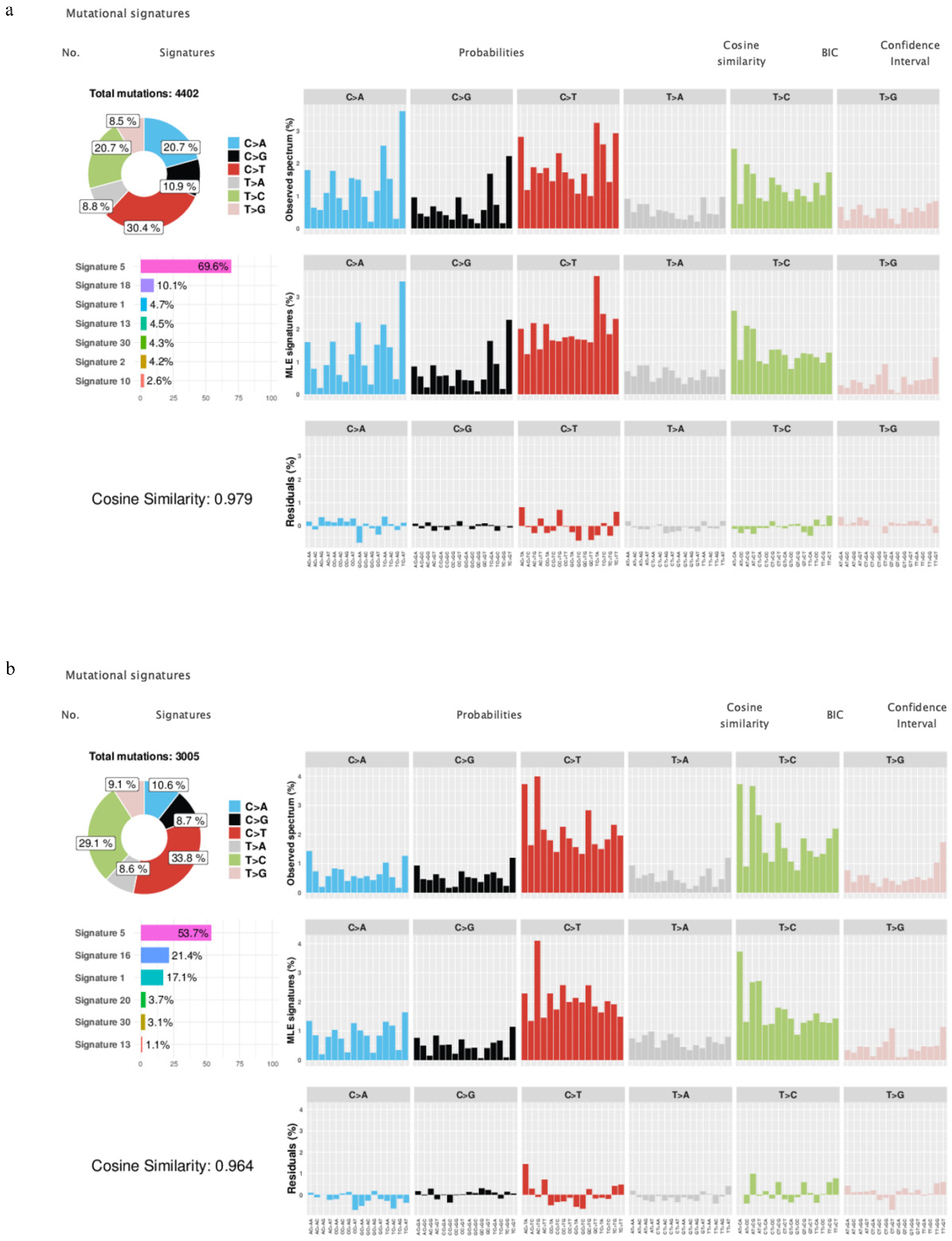
3.3. Mutational Signature
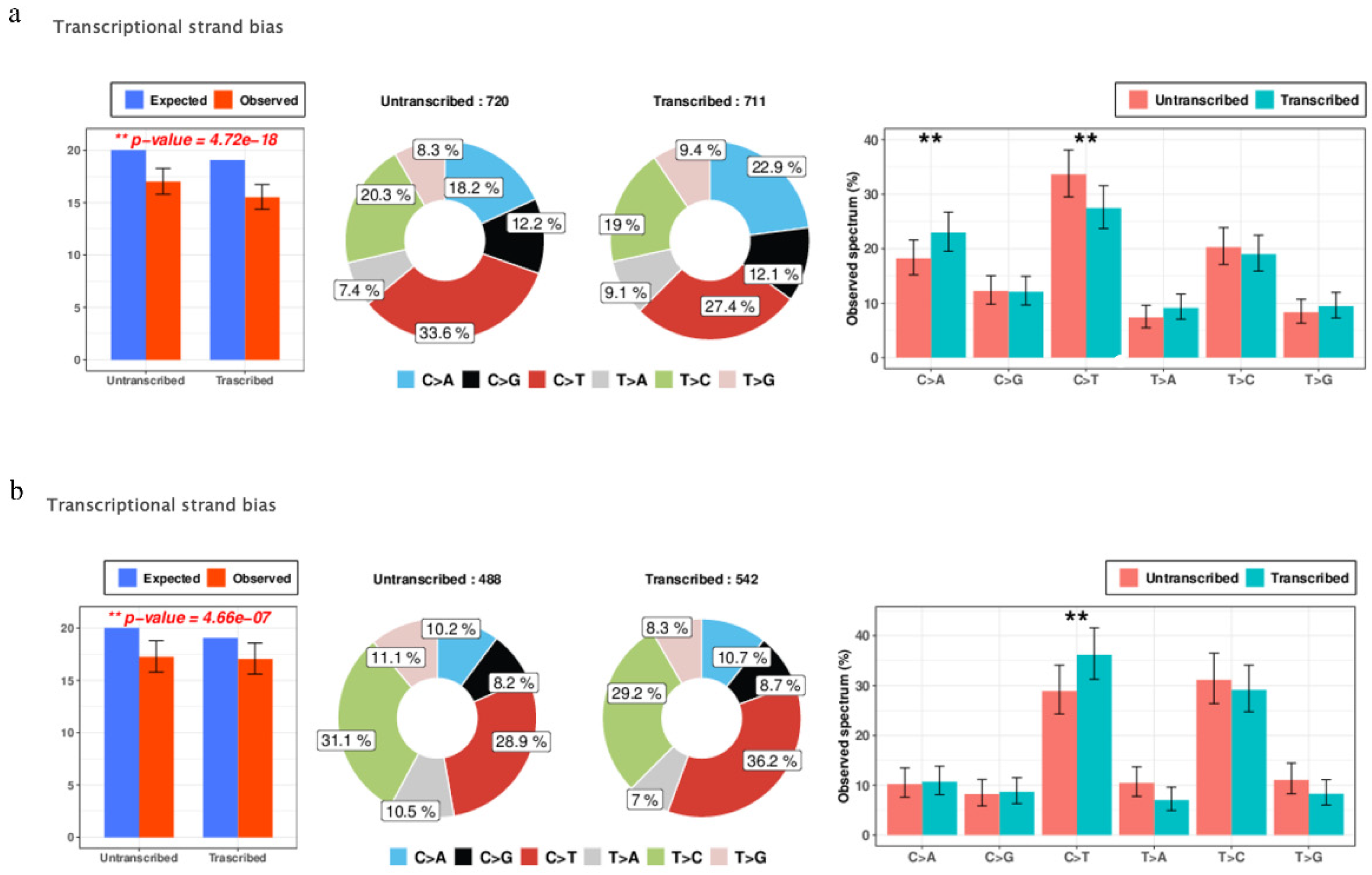
3.4. Transcriptional Strand Bias
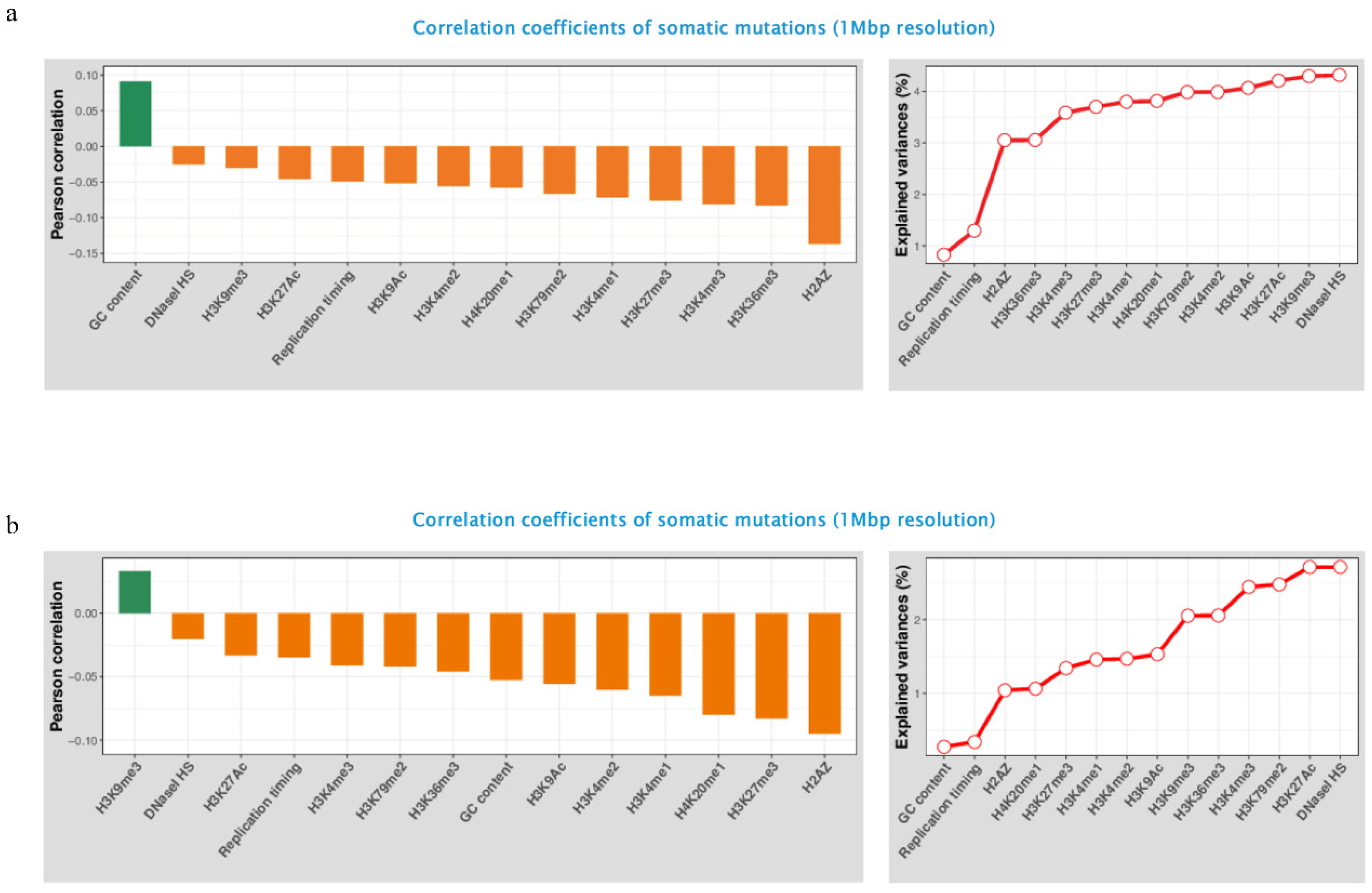
3.5. Genomic and Epigenomic Modification
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Ethics Declarations
Conflicts of Interest
References
- LaTulippe, E.; Satagopan, J.; Smith, A.; Scher, H.; Scardino, P.; Reuter, V.; Gerald, W.L. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002, 62, 4499–4506. [Google Scholar] [PubMed]
- Kyung, S.Y.; Jeong, S.H. Particulate-Matter Related Respiratory Diseases. Tuberc. Respir. Dis. (Seoul) 2020, 83, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiong, L.; Tang, M. Toxicity of inhaled particulate matter on the central nervous system: Neuroinflammation, neuropsychological effects and neurodegenerative disease. J. Appl. Toxicol. 2017, 37, 644–667. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.Q.; FitzGerald, J.M.; Carlsten, C.; Sadatsafavi, M.; Brauer, M. Associations of Ambient Air Pollution with Chronic Obstructive Pulmonary Disease Hospitalization and Mortality. Am. J. Respir. Crit. Care Med. 2013, 187, 721–727. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, J.; Zhang, H.; Shi, C.; Li, G.; Peng, Z.; Ma, J.; Zhou, Y.; Zhang, L. Short-Term Exposure to Ambient Air Pollution and Asthma Mortality. Am. J. Respir. Crit. Care Med. 2019, 200, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Tecer, L.H.; Alagha, O.; Karaca, F.; Tuncel, G.; Eldes, N. Particulate Matter (PM2.5, PM10–2.5, and PM10) and Children’s Hospital Admissions for Asthma and Respiratory Diseases: A Bidirectional Case-Crossover Study. J. Toxicol. Environ. Health Part A 2008, 71, 512–520. [Google Scholar] [CrossRef]
- Johannson, K.A.; Vittinghoff, E.; Lee, K.; Balmes, J.R.; Ji, W.; Kaplan, G.G.; Kim, D.S.; Collard, H.R. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur. Respir. J. 2014, 43, 1124–1131. [Google Scholar] [CrossRef] [Green Version]
- Hamra, G.B.; Guha, N.; Cohen, A.; Laden, F.; Raaschou-Nielsen, O.; Samet, J.M.; Vineis, P.; Forastiere, F.; Saldiva, P.; Yorifuji, T.; et al. Outdoor particulate matter exposure and lung cancer: A systematic review and meta-analysis. Environ. Health Perspect. 2014, 122, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Pan, B.; Wu, J.; Chen, E.; Chen, L. Relationship between exposure to PM2.5 and lung cancer incidence and mortality: A meta-analysis. Oncotarget 2017, 8, 43322–43331. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Lee, A.J.; Lee, J.-K.; Park, J.; Kwon, Y.; Park, S.; Chun, H.; Ju, Y.S.; Hong, D. Mutalisk: A web-based somatic MUTation AnaLyIS toolKit for genomic, transcriptional and epigenomic signatures. Nucleic Acids Res. 2018, 46, W102–W108. [Google Scholar] [CrossRef] [Green Version]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Epstein, D.; Haley, J.D. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin. Exp. Metastasis 2008, 25, 843–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, F.; Tang, D.; Whyatt, R.; Lederman, S.A.; Jedrychowski, W. DNA Damage from Polycyclic Aromatic Hydrocarbons Measured by Benzo[a]pyrene-DNA Adducts in Mothers and Newborns from Northern Manhattan, The World Trade Center Area, Poland, and China. Cancer Epidemiol. Biomark. Prev. 2005, 14, 709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, M.; Autrup, H.; Hertel, O.; Wallin, H.; Knudsen, L.E.; Loft, S. Personal exposure to PM2.5 and biomarkers of DNA damage. Cancer Epidemiol. Biomark. Prev. 2003, 12, 191–196. [Google Scholar]
- Ji, H.; Hershey, G.K.K. Genetic and epigenetic influence on the response to environmental particulate matter. J. Allergy Clin. Immunol. 2012, 129, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, F.; Tang, W.Y.; Herbstman, J.; Tang, D.; Levin, L.; Miller, R.; Ho, S.M. Relation of DNA methylation of 5’-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS ONE 2009, 4, e4488. [Google Scholar] [CrossRef]
- Gilmour, P.S.; Rahman, I.; Donaldson, K.; MacNee, W. Histone acetylation regulates epithelial IL-8 release mediated by oxidative stress from environmental particles. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L533–L540. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Bromberg, P.A.; Samet, J.M. COX-2 expression induced by diesel particles involves chromatin modification and degradation of HDAC1. Am. J. Respir. Cell Mol. Biol. 2007, 37, 232–239. [Google Scholar] [CrossRef]
- Bollati, V.; Marinelli, B.; Apostoli, P.; Bonzini, M.; Nordio, F.; Hoxha, M.; Pegoraro, V.; Motta, V.; Tarantini, L.; Cantone, L.; et al. Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environ. Health Perspect. 2010, 118, 763–768. [Google Scholar] [CrossRef] [Green Version]
- India Project Team of the International Cancer Genome Consortium. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat. Commun. 2013, 4, 2873. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volinia, S.; Druck, T.; Paisie, C.A.; Schrock, M.S.; Huebner, K. The ubiquitous ‘cancer mutational signature’ 5 occurs specifically in cancers with deleted FHIT alleles. Oncotarget 2017, 8, 102199–102211. [Google Scholar] [CrossRef] [Green Version]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell. Mol. Life Sci. 2009, 66, 994–1009. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.D.; Thu, K.L.; Vucic, E.A.; Hubaux, R.; Adonis, M.; Gil, L.; MacAulay, C.; Lam, S.; Lam, W.L. Whole-Genome Sequencing Analysis Identifies a Distinctive Mutational Spectrum in an Arsenic-Related Lung Tumor. J. Thorac. Oncol. 2013, 8, 1451–1455. [Google Scholar] [CrossRef] [Green Version]
- Genchi, G.; Carocci, A.; Lauria, G.; Sinicropi, M.S.; Catalano, A. Nickel: Human Health and Environmental Toxicology. Int. J. Environ. Res. Public Health 2020, 17, 679. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liang, Y.; Guan, B.; Shi, Y.; Gong, Y.; Li, J.; Kong, W.; Liu, J.; Fang, D.; Liu, L.; et al. Aristolochic acid mutational signature defines the low-risk subtype in upper tract urothelial carcinoma. Theranostics 2020, 10, 4323–4333. [Google Scholar] [CrossRef]
- Wei, R.; Li, P.; He, F.; Wei, G.; Zhou, Z.; Su, Z.; Ni, T. Comprehensive analysis reveals distinct mutational signature and its mechanistic insights of alcohol consumption in human cancers. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Kucab, J.E.; Phillips, D.H.; Arlt, V.M. Linking environmental carcinogen exposure to TP53 mutations in human tumours using the human TP53 knock-in (Hupki) mouse model. FEBS J. 2010, 277, 2567–2583. [Google Scholar] [CrossRef] [PubMed]
- vom Brocke, J.; Krais, A.; Whibley, C.; Hollstein, M.C.; Schmeiser, H.H. The carcinogenic air pollutant 3-nitrobenzanthrone induces GC to TA transversion mutations in human p53 sequences. Mutagenesis 2009, 24, 17–23. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Wang, J.; Bao, L.; Wang, L.; Huo, J.; Wang, X. Global analysis of chromosome 1 genes among patients with lung adenocarcinoma, squamous carcinoma, large-cell carcinoma, small-cell carcinoma, or non-cancer. Cancer Metastasis Rev. 2015, 34, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Lockwood, W.W.; Buys, T.P.; Chari, R.; Coe, B.P.; Lam, S.; Lam, W.L. Integrative genomic and gene expression analysis of chromosome 7 identified novel oncogene loci in non-small cell lung cancer. Genome 2008, 51, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Wan, H.; Dong, N.; Bao, L.; Sun, X.; Xu, M.; Wang, X. Global analyses of subtype- or stage-specific genes on chromosome 7 in patients with lung cancer. Cancer Metastasis Rev. 2015, 34, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Cadelis, G.; Tourres, R.; Molinie, J. Short-term effects of the particulate pollutants contained in Saharan dust on the visits of children to the emergency department due to asthmatic conditions in Guadeloupe (French Archipelago of the Caribbean). PLoS ONE 2014, 9, e91136. [Google Scholar] [CrossRef] [Green Version]
- Downs, S.H.; Schindler, C.; Liu, L.J.S.; Keidel, D.; Bayer-Oglesby, L.; Brutsche, M.H.; Gerbase, M.W.; Keller, R.; Künzli, N.; Leuenberger, P.; et al. Reduced Exposure to PM10 and Attenuated Age-Related Decline in Lung Function. N. Engl. J. Med. 2007, 357, 2338–2347. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.J.; Ku, G.W.; Lee, S.Y.; Kang, D.; Hwang, W.J.; Jeong, I.B.; Kwon, S.J.; Kang, J.; Son, J.W. Analysis of Single Nucleotide Variants (SNVs) Induced by Exposure to PM10 in Lung Epithelial Cells Using Whole Genome Sequencing. Int. J. Environ. Res. Public Health 2021, 18, 1046. https://doi.org/10.3390/ijerph18031046
Park SJ, Ku GW, Lee SY, Kang D, Hwang WJ, Jeong IB, Kwon SJ, Kang J, Son JW. Analysis of Single Nucleotide Variants (SNVs) Induced by Exposure to PM10 in Lung Epithelial Cells Using Whole Genome Sequencing. International Journal of Environmental Research and Public Health. 2021; 18(3):1046. https://doi.org/10.3390/ijerph18031046
Chicago/Turabian StylePark, Se Jin, Gwan Woo Ku, Su Yel Lee, Daeun Kang, Wan Jin Hwang, In Beom Jeong, Sun Jung Kwon, Jaeku Kang, and Ji Woong Son. 2021. "Analysis of Single Nucleotide Variants (SNVs) Induced by Exposure to PM10 in Lung Epithelial Cells Using Whole Genome Sequencing" International Journal of Environmental Research and Public Health 18, no. 3: 1046. https://doi.org/10.3390/ijerph18031046