
Handling Poor Accrual in Pediatric Trials: A Simulation Study Using a Bayesian Approach

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Motivating Example
2.2. Simulation Plan
- Data generation hypotheses.
- Analysis of simulated data.
- Presentation of the results of simulations.
2.3. Data Generation Hypotheses
2.3.1. Simulation Scenarios
2.3.2. Data Generation within Scenarios
2.4. Analysis of the Simulated Data
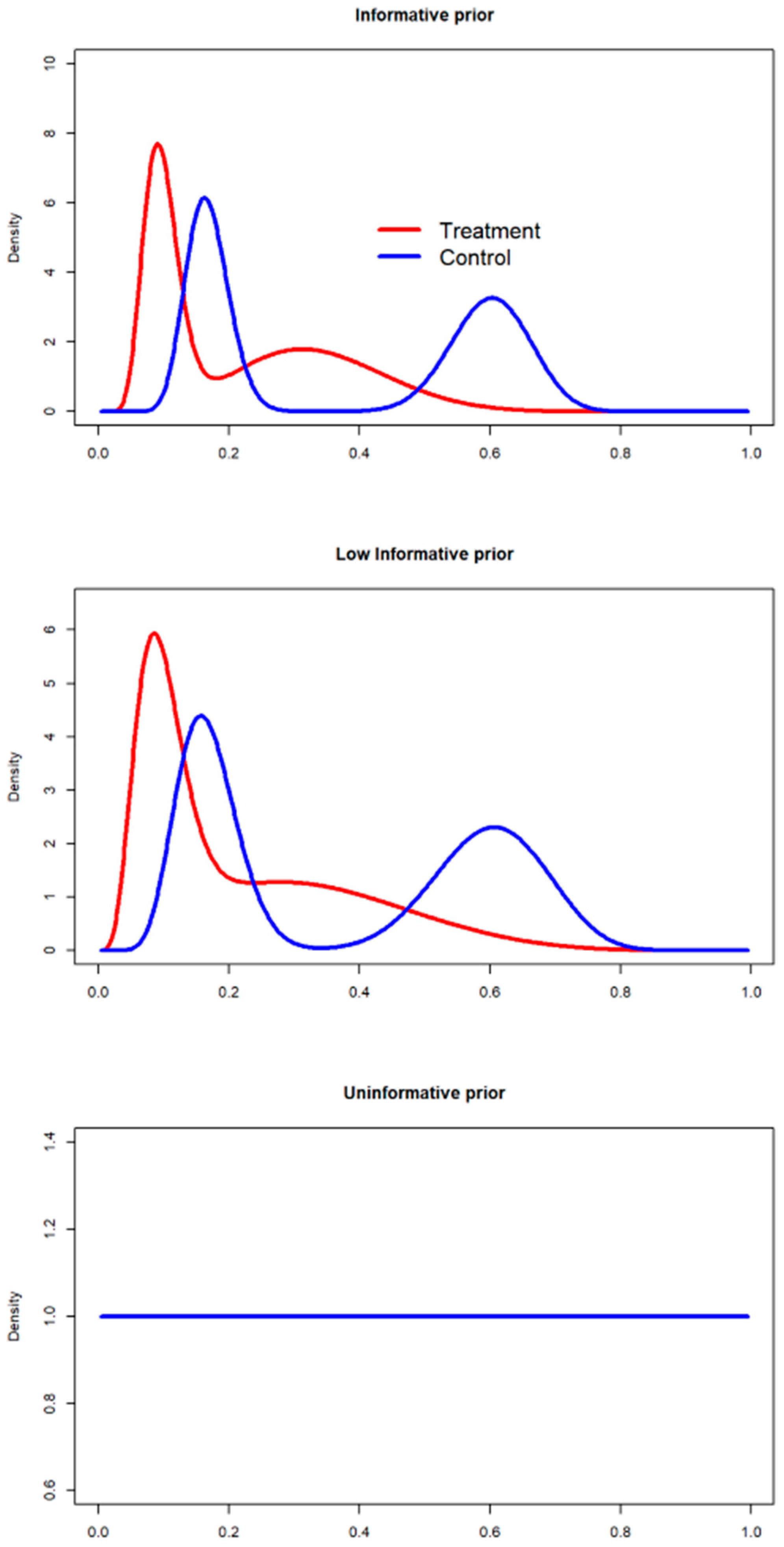
2.4.1. Prior Definition
- Huang et al. [38] reported probabilities of scarring of and in the treatment and control arms, respectively. Considering this information, the informative beta prior can be derived as:
- Shaikh et al. [39] reported, instead, probabilities of scarring of (12|123) and (22|131) in the treatment and control arms, respectively. Considering this information, the informative beta prior can be derived as:
- For the treatment arm, the beta mixture is defined as:The expected value for the mixture random variable is, for the treatment arm, a weighted mean of the expectations over the mixture components:If we denote the beta shape and , respectively for the Huang and Shaikh studies, and and the scales for the considered studies, the mixture expected value may be computed as:If we assume an equal weight value , .
- The mixture variance is given by:where the variances of the mixture components are:Equal weight was assumed for the components of the mixture, therefore, , , and SD[= 0.08.
- For the treatment arm, the mixture is defined as:
2.4.2. Discounting the Priors: The Power Prior Approach
- If = 0, the data provided by the literature are not considered, indicating a 100% discount of the historical information. According to this scenario, the prior is an uninformative distribution.
- If = 1, then all of the information provided by the literature is considered in setting the prior, indicating a 0% discount of the historical data.
- Power prior without discounting (informative, = 1). A beta informative prior was derived considering the number of successes and failures found in the literature [42], as defined in the method section.
- Power prior 50% discounting (low-informative, = 0.5). The beta prior with a 50% discount, defined in the literature as a substantial-moderate discounting factor [43], was defined based on the beta parameters comprising the mixture of priors specified in the informative scenario.
- Power prior 100% discounting (uninformative, = 0). A mixture of priors was defined.
Effective Sample Size (ESS) Calculation
2.4.3. Posterior Estimation
- A first resampling of the proportion of scarring from , which is the posterior distribution for the treatment group.
- A second resampling of from .
- The posterior distribution for the parameter related to the difference in proportions was obtained by calculating from the distributions previously resampled [47].
2.4.4. Convergence Assessment
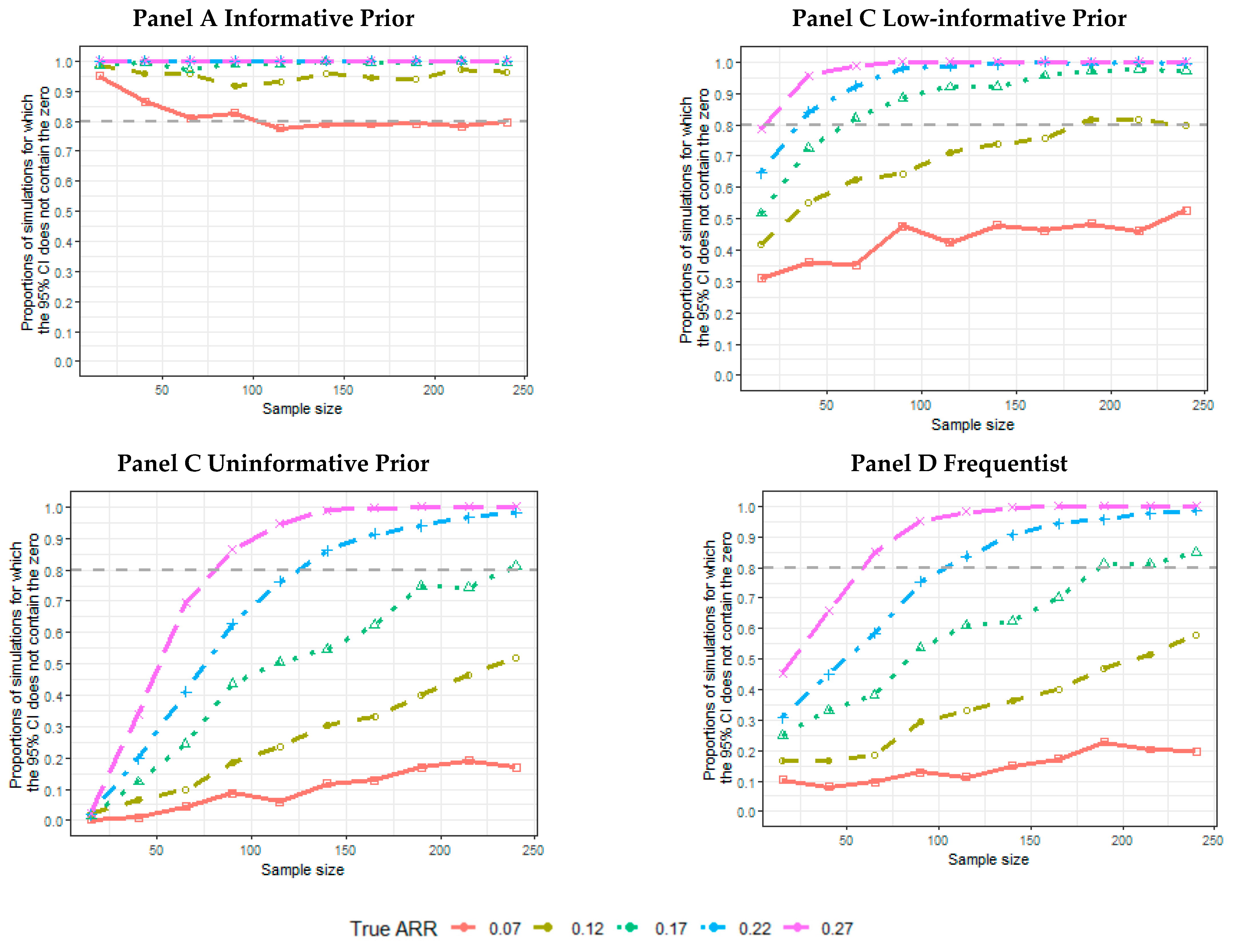
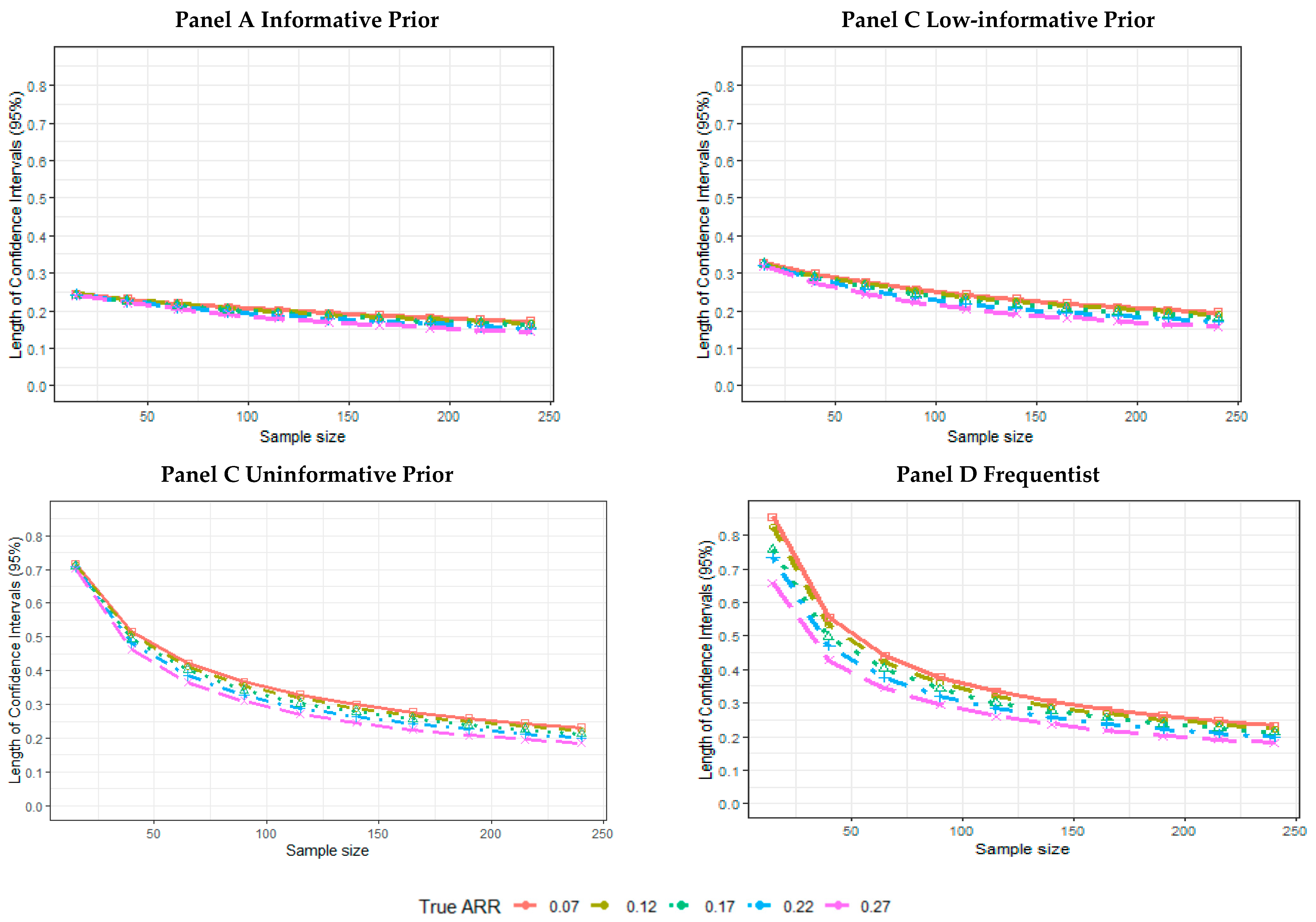
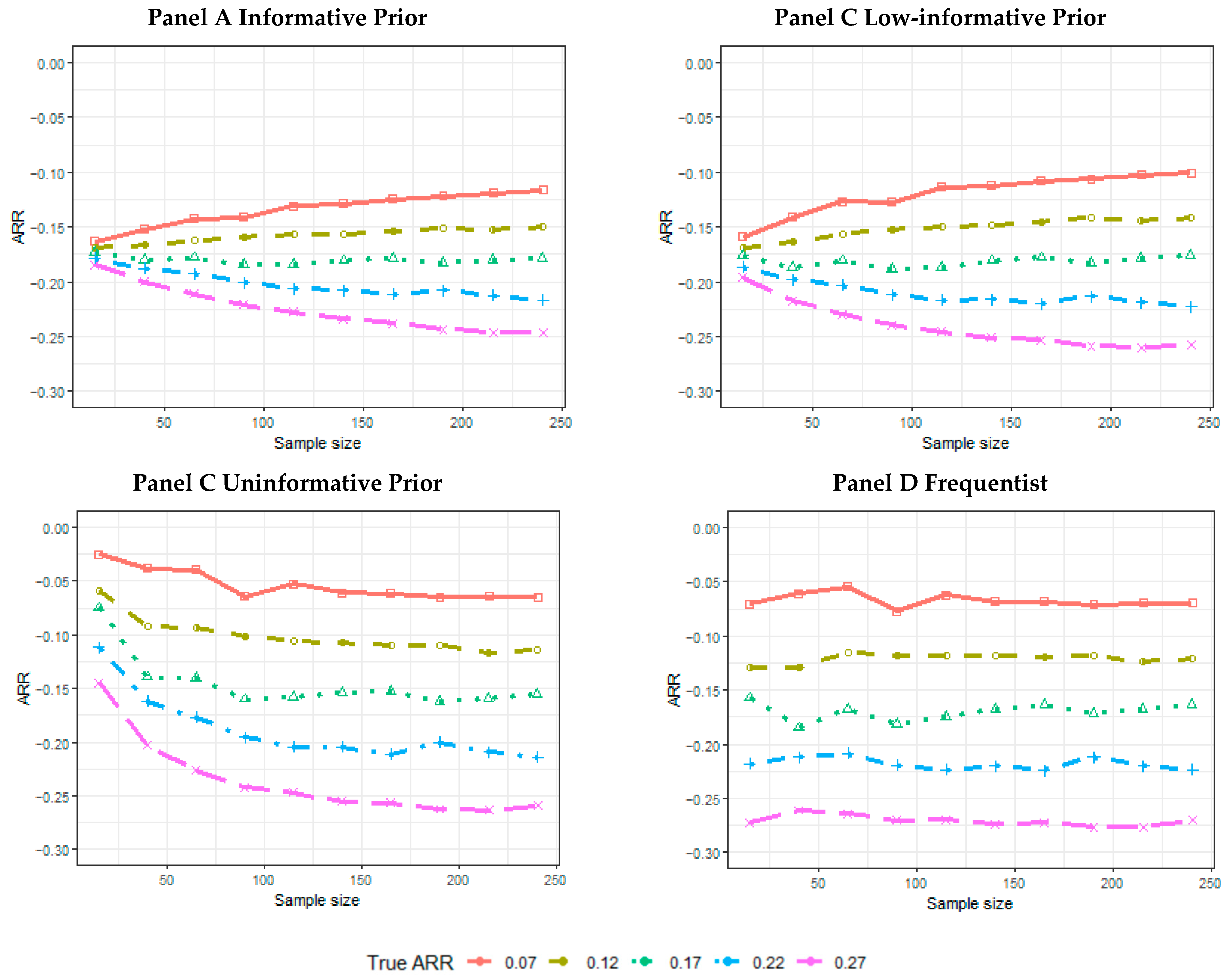
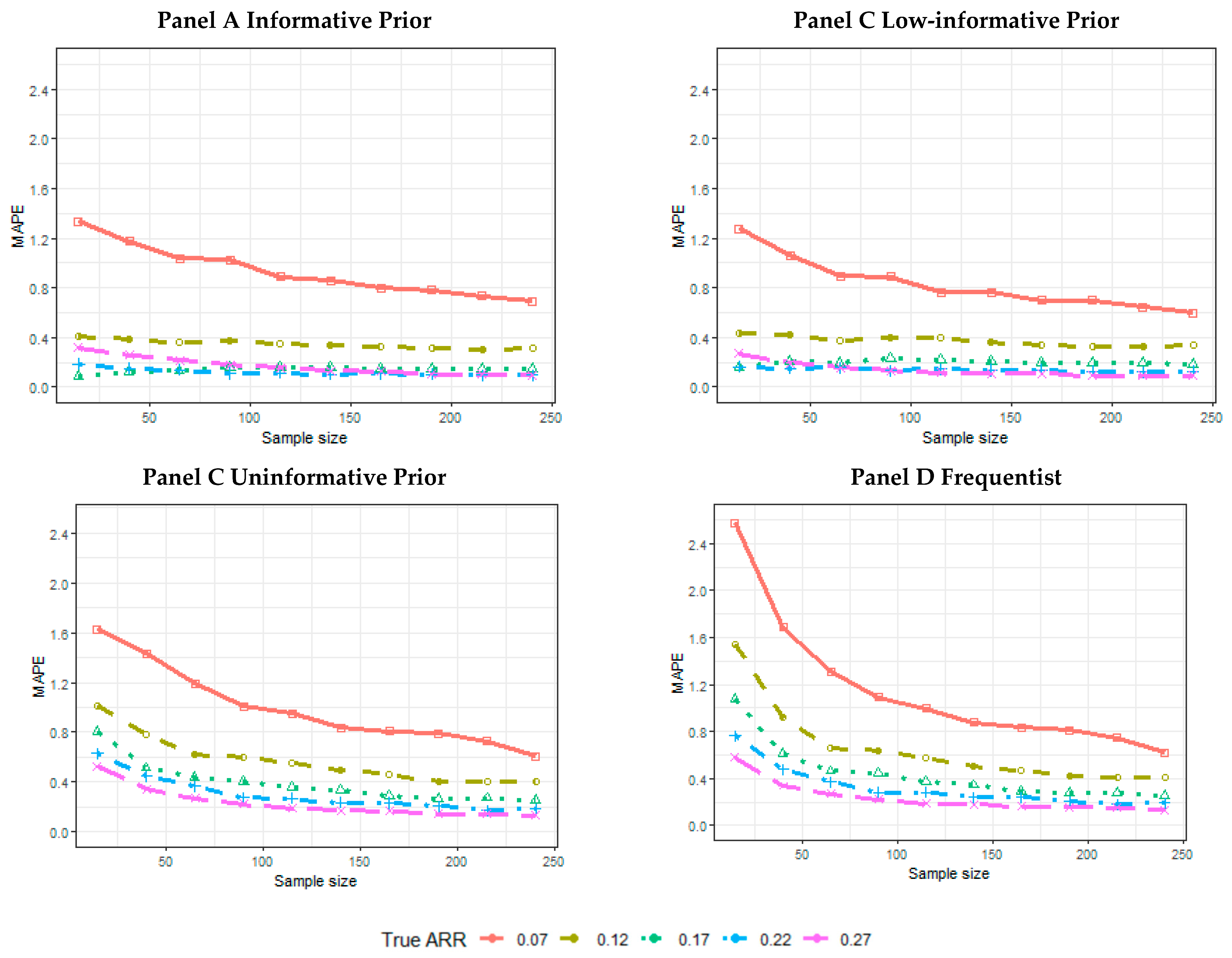
2.5. Results of the Simulations
- The proportion of the 5000 simulated trials for which the credibility intervals (CIs), or confidence intervals, for a frequentist analysis do not contain an ARR equal to 0. The proportion of intervals not containing the 0 and containing the data generator ARR was also calculated.
- The mean length across 5000 simulated trials of the CI.
- The mean of the posterior median estimate across 5000 simulated trials or the mean of the point-estimated ARR across 5000 simulated trials for the frequentist analysis.
- The mean absolute percentage error (MAPE):is the true treatment effect considered to generate the data; is the estimated treatment effect (posterior median, or point estimate, for the frequentist analysis) achieved for each simulation t within the n = 5000 simulated trials.
3. Results
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kadam, R.A.; Borde, S.U.; Madas, S.A.; Salvi, S.S.; Limaye, S.S. Challenges in Recruitment and Retention of Clinical Trial Subjects. Perspect. Clin. Res. 2016, 7, 137–143. [Google Scholar] [CrossRef]
- Pak, T.R.; Rodriguez, M.; Roth, F.P. Why Clinical Trials Are Terminated. bioRxiv 2015, bioRxiv:021543. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.J.; Tse, T.; DiPiazza, K.; Zarin, D.A. Terminated Trials in the ClinicalTrials.gov Results Database: Evaluation of Availability of Primary Outcome Data and Reasons for Termination. PLoS ONE 2015, 10, e0127242. [Google Scholar] [CrossRef] [Green Version]
- Rimel, B. Clinical Trial Accrual: Obstacles and Opportunities. Front. Oncol. 2016, 6, 103. [Google Scholar] [CrossRef] [Green Version]
- Mannel, R.S.; Moore, K. Research: An Event or an Environment? Gynecol. Oncol. 2014, 134, 441–442. [Google Scholar] [CrossRef]
- Stensland, K.D.; McBride, R.B.; Latif, A.; Wisnivesky, J.; Hendricks, R.; Roper, N.; Boffetta, P.; Hall, S.J.; Oh, W.K.; Galsky, M.D. Adult Cancer Clinical Trials That Fail to Complete: An Epidemic? J. Natl. Cancer Inst. 2014, 106, dju229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldi, I.; Lanera, C.; Berchialla, P.; Gregori, D. Early Termination of Cardiovascular Trials as a Consequence of Poor Accrual: Analysis of ClinicalTrials.gov 2006–2015. BMJ Open 2017, 7, e013482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pica, N.; Bourgeois, F. Discontinuation and Nonpublication of Randomized Clinical Trials Conducted in Children. Pediatrics 2016, e20160223. [Google Scholar] [CrossRef] [Green Version]
- Baiardi, P.; Giaquinto, C.; Girotto, S.; Manfredi, C.; Ceci, A. Innovative Study Design for Paediatric Clinical Trials. Eur. J. Clin. Pharmacol. 2011, 67, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, R.G.; Gamel, B.; Bloom, D.; Bradley, J.; Jafri, H.S.; Hinton, D.; Nambiar, S.; Wheeler, C.; Tiernan, R.; Smith, P.B.; et al. Parents’ Perceived Obstacles to Pediatric Clinical Trial Participation: Findings from the Clinical Trials Transformation Initiative. Contemp. Clin. Trials Commun. 2018, 9, 33–39. [Google Scholar] [CrossRef]
- Billingham, L.; Malottki, K.; Steven, N. Small Sample Sizes in Clinical Trials: A Statistician’s Perspective. Clin. Investig. 2012, 2, 655–657. [Google Scholar] [CrossRef]
- Kitterman, D.R.; Cheng, S.K.; Dilts, D.M.; Orwoll, E.S. The Prevalence and Economic Impact of Low-Enrolling Clinical Studies at an Academic Medical Center. Acad. Med. J. Assoc. Am. Med Coll. 2011, 86, 1360–1366. [Google Scholar] [CrossRef] [Green Version]
- Joseph, P.D.; Craig, J.C.; Caldwell, P.H. Clinical Trials in Children. Br. J. Clin. Pharmacol. 2015, 79, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Huff, R.A.; Maca, J.D.; Puri, M.; Seltzer, E.W. Enhancing Pediatric Clinical Trial Feasibility through the Use of Bayesian Statistics. Pediatric Res. 2017, 82, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICH Topic E 11. Clinical Investigation of Medicinal Products in the Paediatric Population. In Note for Guidance on Clinical Investigation of Medicinal Products in the Paediatric Population (CPMP/ICH/2711/99); European Medicines Agency: London, UK, 2001. [Google Scholar]
- European Medicines Agency. Guideline on the Requirements for Clinical Documentation for Orally Inhaled Products (OIP) Including the Requirements for Demonstration of Therapeutic Equivalence between Two Inhaled Products for Use in the Treatment of Asthma and Chronic Obstructive Pulmonary Disease (COPD) in Adults and for Use in the Treatment of Asthma in Children and Adolescents; European Medicines Agency: London, UK, 2009. [Google Scholar]
- Committee for Medicinal Products for Human Use. Guideline on the Clinical Development of Medicinal Products for the Treatment of Cystic Fibrosis; European Medicines Agency: London, UK, 2009. [Google Scholar]
- Committee for Medicinal Products for Human Use. Note for Guidance on Evaluation of Anticancer Medicinal Products in Man; The European Agency for the Evaluation of Medical Products: London, UK, 1996. [Google Scholar]
- O’Hagan, A. Chapter 6: Bayesian Statistics: Principles and Benefits. In Handbook of Probability: Theory and Applications; Rudas, T., Ed.; Sage: Thousand Oaks, CA, USA, 2008; pp. 31–45. [Google Scholar]
- Azzolina, D.; Berchialla, P.; Gregori, D.; Baldi, I. Prior Elicitation for Use in Clinical Trial Design and Analysis: A Literature Review. Int. J. Environ. Res. Public Health 2021, 18, 1833. [Google Scholar] [CrossRef]
- Gajewski, B.J.; Simon, S.D.; Carlson, S.E. Predicting Accrual in Clinical Trials with Bayesian Posterior Predictive Distributions. Stat. Med. 2008, 27, 2328–2340. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, A. Eliciting Expert Beliefs in Substantial Practical Applications. J. R. Stat. Soc. Ser. D Stat. [CD-ROM P55-68]. 1998, 47, 21–35. [Google Scholar]
- Lilford, R.J.; Thornton, J.; Braunholtz, D. Clinical Trials and Rare Diseases: A Way out of a Conundrum. BMJ 1995, 311, 1621–1625. [Google Scholar] [CrossRef] [Green Version]
- Quintana, M.; Viele, K.; Lewis, R.J. Bayesian Analysis: Using Prior Information to Interpret the Results of Clinical Trials. Jama 2017, 318, 1605–1606. [Google Scholar] [CrossRef]
- Office of the Commissioner; Office of Clinical Policy and Programs. Guidance for the Use of Bayesian Statistics in Medical Device Clinical Trials; CDRH: Rockville, MD, USA; CBER: Silver Spring, MD, USA, 2010. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-use-bayesian-statistics-medical-device-clinical-trials (accessed on 2 February 2021).
- Gelman, A. Prior Distribution. Chapter 4: Statistical Theory and Methods. In Encyclopedia of Environmetrics; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- De Santis, F. Power Priors and Their Use in Clinical Trials. Am. Stat. 2006, 60, 122–129. [Google Scholar] [CrossRef]
- Ibrahim, J.G.; Chen, M.-H. Power Prior Distributions for Regression Models. Stat. Sci. 2000, 15, 46–60. [Google Scholar]
- Psioda, M.A.; Ibrahim, J.G. Bayesian Clinical Trial Design Using Historical Data That Inform the Treatment Effect. Biostat 2019, 20, 400–415. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, J.G.; Chen, M.-H.; Gwon, Y.; Chen, F. The Power Prior: Theory and Applications. Stat. Med. 2015, 34, 3724–3749. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, B.P.; Carlin, B.P.; Mandrekar, S.J.; Sargent, D.J. Hierarchical Commensurate and Power Prior Models for Adaptive Incorporation of Historical Information in Clinical Trials. Biometrics 2011, 67, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.F. A Dynamic Power Prior for Borrowing Historical Data in Noninferiority Trials with Binary Endpoint. Pharm. Stat. 2018, 17, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Nikolakopoulos, S.; van der Tweel, I.; Roes, K.C.B. Dynamic Borrowing through Empirical Power Priors That Control Type I Error: Dynamic Borrowing with Type I Error Control. Biom 2018, 74, 874–880. [Google Scholar] [CrossRef]
- Ollier, A.; Morita, S.; Ursino, M.; Zohar, S. An Adaptive Power Prior for Sequential Clinical Trials—Application to Bridging Studies. Stat. Methods Med Res. 2020, 29, 2282–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidli, H.; Gsteiger, S.; Roychoudhury, S.; O’Hagan, A.; Spiegelhalter, D.; Neuenschwander, B. Robust Meta-Analytic-Predictive Priors in Clinical Trials with Historical Control Information: Robust Meta-Analytic-Predictive Priors. Biom 2014, 70, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Chuang-Stein, C. An Application of the Beta-Binomial Model to Combine and Monitor Medical Event Rates in Clinical Trials. Drug Inf. J. 1993, 27, 515–523. [Google Scholar] [CrossRef]
- Zaslavsky, B.G. Bayes Models of Clinical Trials with Dichotomous Outcomes and Sample Size Determination. Stat. Biopharm. Res. 2009, 1, 149–158. [Google Scholar] [CrossRef]
- Huang, Y.C.; Lin, Y.C.; Wei, C.F.; Deng, W.L.; Huang, H.C. The Pathogenicity Factor HrpF Interacts with HrpA and HrpG to Modulate Type III Secretion System (T3SS) Function and T3ss Expression in Pseudomonas Syringae Pv. Averrhoi. Mol. Plant Pathol. 2016, 17, 1080–1094. [Google Scholar] [CrossRef]
- Shaikh, N.; Shope, T.R.; Hoberman, A.; Muniz, G.B.; Bhatnagar, S.; Nowalk, A.; Hickey, R.W.; Michaels, M.G.; Kearney, D.; Rockette, H.E.; et al. Corticosteroids to Prevent Kidney Scarring in Children with a Febrile Urinary Tract Infection: A Randomized Trial. Pediatr. Nephrol. 2020, 35, 2113–2120. [Google Scholar] [CrossRef]
- Lehoczky, J.P. Distributions, Statistical: Special and Continuous. In International Encyclopedia of the Social & Behavioral Sciences; Elsevier: Amsterdam, The Netherlands, 2001; pp. 3787–3793. ISBN 978-0-08-043076-8. [Google Scholar]
- Wilcox, R.R. A Review of the Beta-Binomial Model and Its Extensions. J. Educ. Stat. 1981, 6, 3. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; Chen, M.-J.; Chiu, N.-T.; Chou, H.-H.; Lin, K.-Y.; Chiou, Y.-Y. Adjunctive Oral Methylprednisolone in Pediatric Acute Pyelonephritis Alleviates Renal Scarring. Pediatrics 2011, peds-2010. [Google Scholar] [CrossRef]
- De Santis, F. Using Historical Data for Bayesian Sample Size Determination. J. R. Stat. Soc. Ser. A 2007, 170, 95–113. [Google Scholar] [CrossRef]
- Weber, S. RBesT: R Bayesian Evidence Synthesis Tools, Version 1.6-1. Available online: https://cran.r-project.org/web/packages/RBesT/index.html (accessed on 2 February 2020).
- Albert, J. Bayesian Computation with R; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2009; ISBN 0-387-92298-9. [Google Scholar]
- Kawasaki, Y.; Shimokawa, A.; Miyaoka, E. Comparison of Three Calculation Methods for a Bayesian Inference of P (Π1 > Π2). J. Mod. Appl. Stat. Methods 2013, 12, 256–268. [Google Scholar] [CrossRef]
- Barry, J. Doing Bayesian Data Analysis: A Tutorial with R and BUGS. Eur. J. Psychol. 2011, 7, 778–779. [Google Scholar] [CrossRef]
- Lunn, D.; Spiegelhalter, D.; Thomas, A.; Best, N. The BUGS Project: Evolution, Critique and Future Directions. Stat. Med. 2009, 28, 3049–3067. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Geweke, J. Evaluating the Accuracy of Sampling-Based Approaches to the Calculation of Posterior Moments; Federal Reserve Bank of Minneapolis, Research Department: Minneapolis, MN, USA, 1991; Volume 196. [Google Scholar]
- Bavdekar, S.B. Pediatric Clinical Trials. Perspect. Clin. Res. 2013, 4, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.; Kurz, R. Practical and Ethical Issues in Pediatric Clinical Trials. Appl. Clin. Trials 2003, 12, 41–45. [Google Scholar]
- Pedroza, C.; Han, W.; Thanh Truong, V.T.; Green, C.; Tyson, J.E. Performance of Informative Priors Skeptical of Large Treatment Effects in Clinical Trials: A Simulation Study. Stat. Methods Med. Res. 2018, 27, 79–96. [Google Scholar] [CrossRef] [PubMed]
- McNeish, D. On Using Bayesian Methods to Address Small Sample Problems. Struct. Equ. Modeling Multidiscip. J. 2016, 23, 750–773. [Google Scholar] [CrossRef]
- Depaoli, S. Mixture Class Recovery in GMM under Varying Degrees of Class Separation: Frequentist versus Bayesian Estimation. Psychol. Methods 2013, 18, 186–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegelhalter, D.J.; Freedman, L.S.; Parmar, M.K.B. Bayesian Approaches to Randomized Trials. J. R. Stat. Soc. Ser. A 1994, 157, 357. [Google Scholar] [CrossRef]
- Albers, C.J.; Kiers, H.A.L.; van Ravenzwaaij, D. Credible Confidence: A Pragmatic View on the Frequentist vs. Bayesian Debate. Collabra Psychol. 2018, 4, 31. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scenario | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 |
| Sample size | 15 | 40 | 65 | 90 | 115 | 140 | 165 | 190 | 215 | 240 | 15 | 40 | 65 | 90 | 115 | 140 | 165 | 190 | 215 | 240 | 15 | 40 | 65 | 90 | 115 |
| True ARR | 0.07 | 0.07 | 0.07 | 0.07 | 0.07 | 0.07 | 0.07 | 0.07 | 0.07 | 0.07 | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 |
| Scenario | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | 47 | 48 | 49 | 50 |
| Sample size | 140 | 165 | 190 | 215 | 240 | 15 | 40 | 65 | 90 | 115 | 140 | 165 | 190 | 215 | 240 | 15 | 40 | 65 | 90 | 115 | 140 | 165 | 190 | 215 | 240 |
| True ARR | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 | 0.22 | 0.22 | 0.22 | 0.22 | 0.22 | 0.22 | 0.22 | 0.22 | 0.22 | 0.22 | 0.27 | 0.27 | 0.27 | 0.27 | 0.27 | 0.27 | 0.27 | 0.27 | 0.27 | 0.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzolina, D.; Lorenzoni, G.; Bressan, S.; Da Dalt, L.; Baldi, I.; Gregori, D. Handling Poor Accrual in Pediatric Trials: A Simulation Study Using a Bayesian Approach. Int. J. Environ. Res. Public Health 2021, 18, 2095. https://doi.org/10.3390/ijerph18042095
Azzolina D, Lorenzoni G, Bressan S, Da Dalt L, Baldi I, Gregori D. Handling Poor Accrual in Pediatric Trials: A Simulation Study Using a Bayesian Approach. International Journal of Environmental Research and Public Health. 2021; 18(4):2095. https://doi.org/10.3390/ijerph18042095
Chicago/Turabian StyleAzzolina, Danila, Giulia Lorenzoni, Silvia Bressan, Liviana Da Dalt, Ileana Baldi, and Dario Gregori. 2021. "Handling Poor Accrual in Pediatric Trials: A Simulation Study Using a Bayesian Approach" International Journal of Environmental Research and Public Health 18, no. 4: 2095. https://doi.org/10.3390/ijerph18042095