
Clinical Spectrum Associated with Wolfram Syndrome Type 1 and Type 2: A Review on Genotype–Phenotype Correlations



Abstract
:1. Introduction
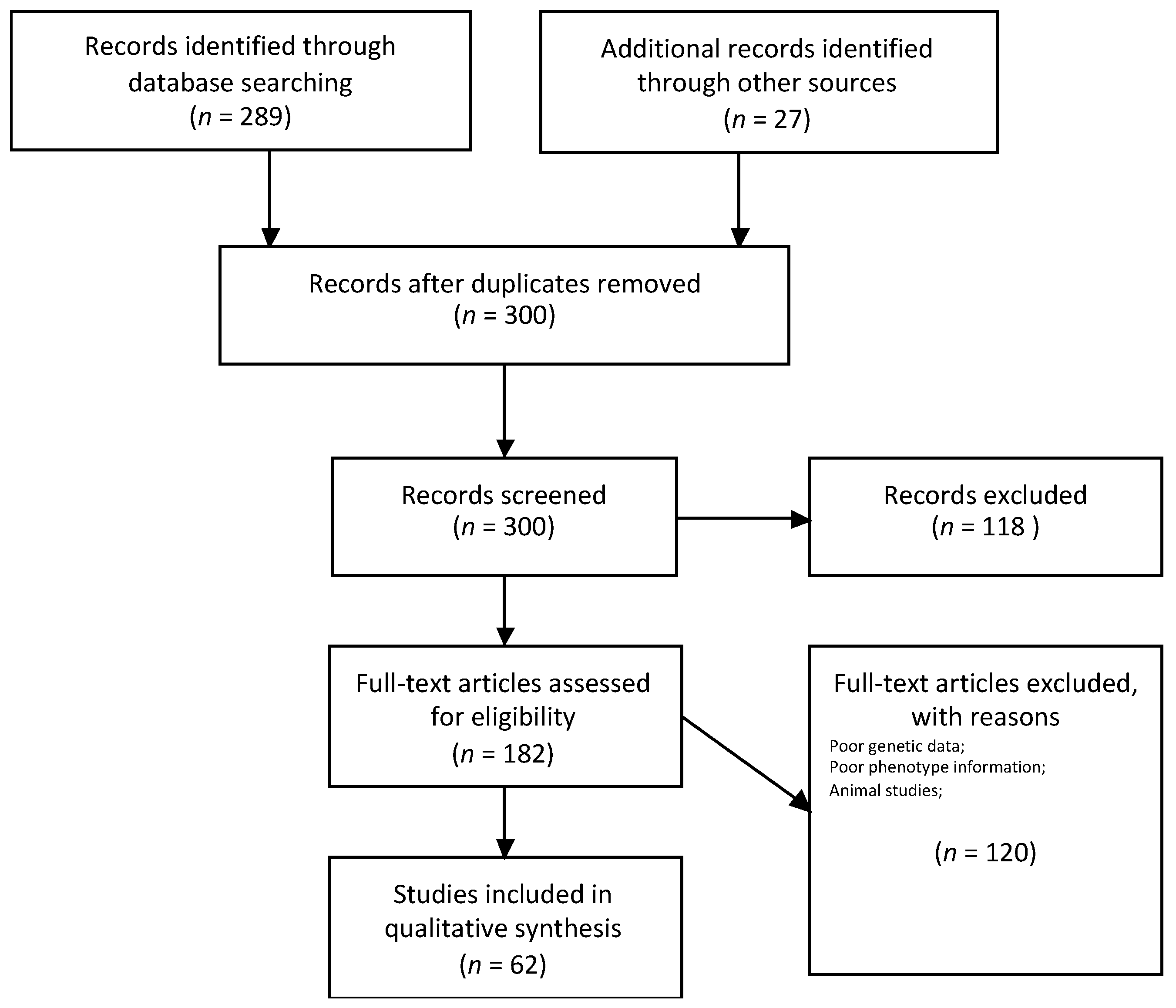
2. Methods
3. Epidemiology
4. Clinical Findings and Treatment
Treatment Perspective
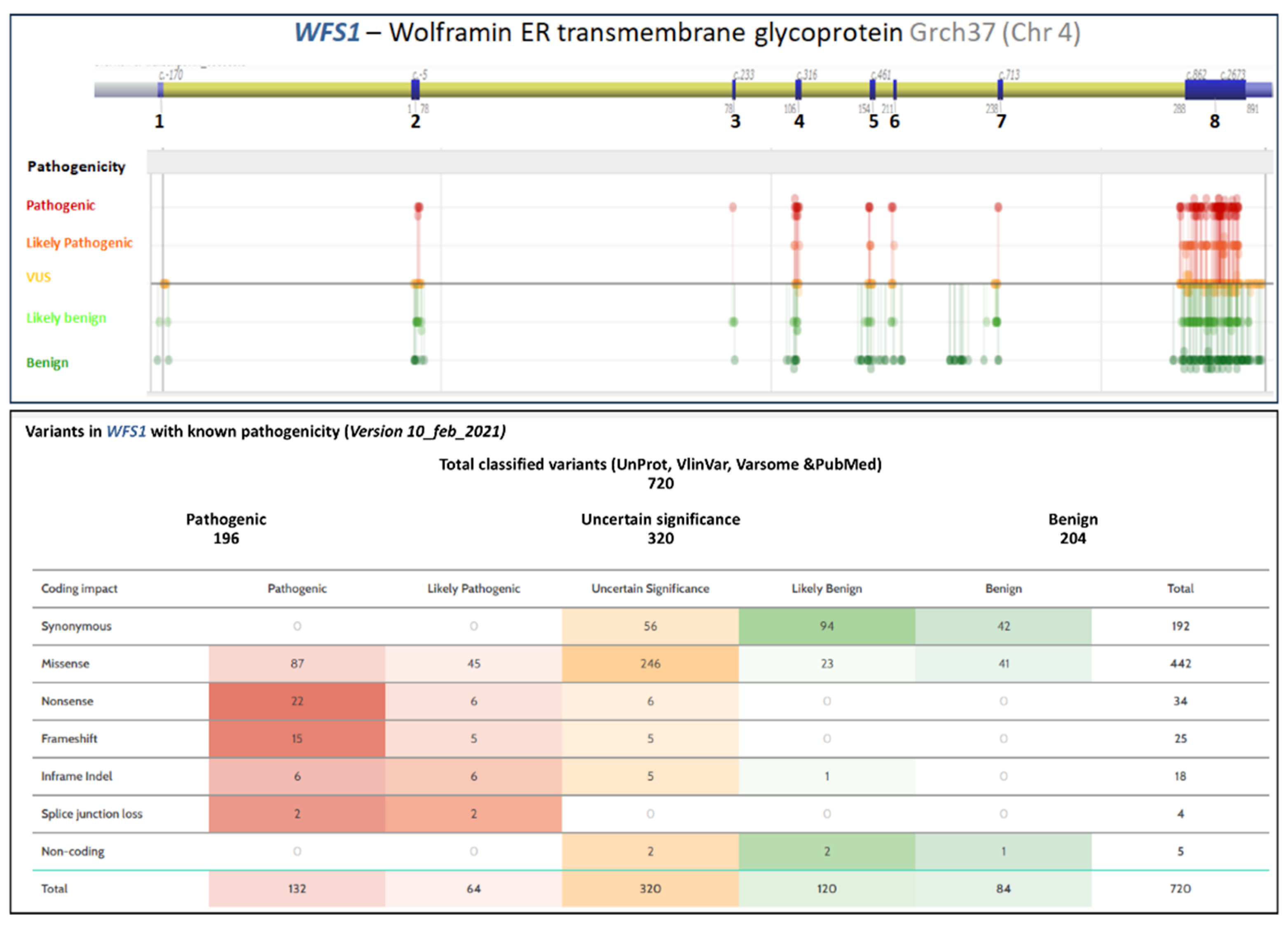
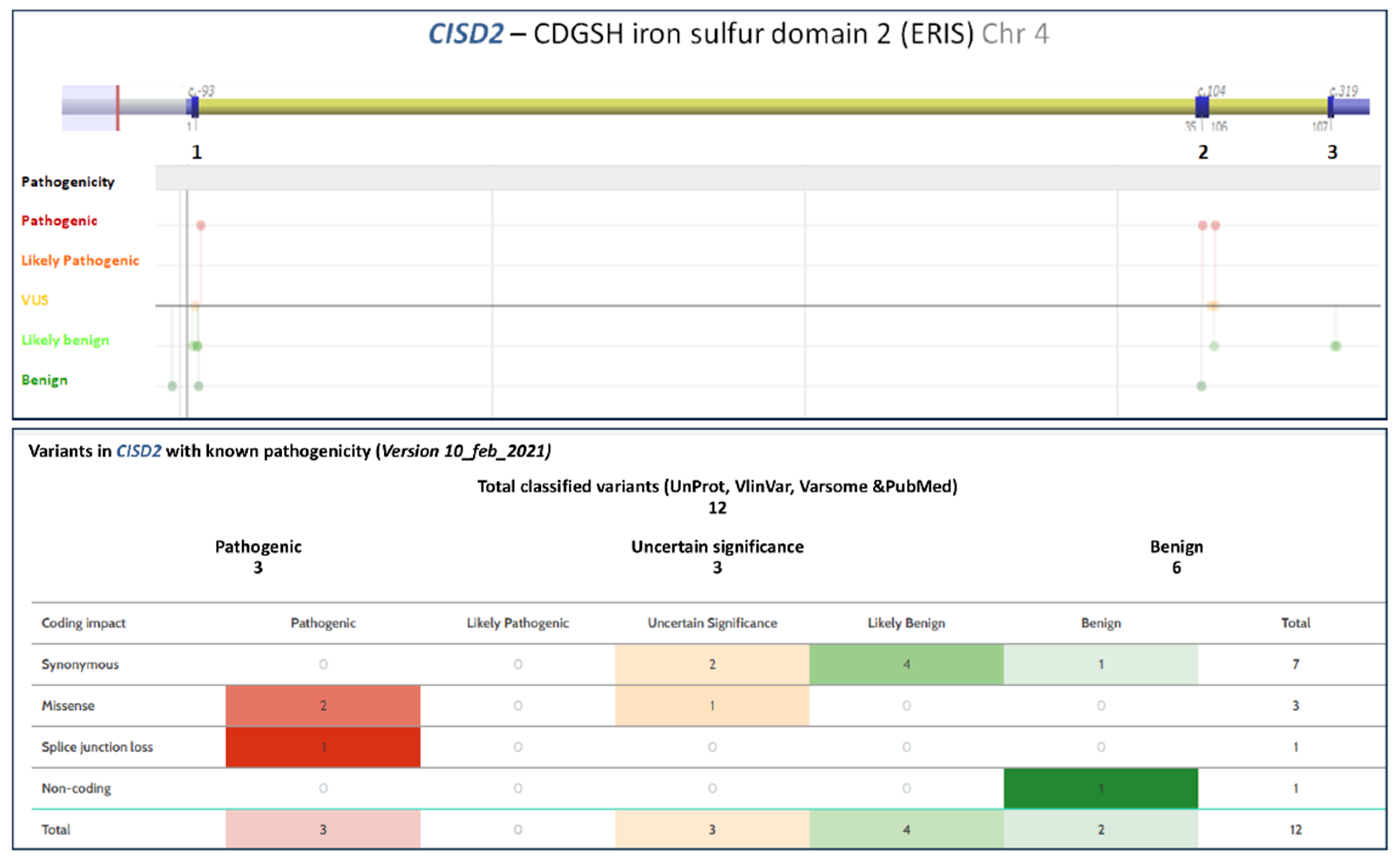
5. Molecular Genetics
5.1. Wolfram Syndrome Type 1 Genotype–Phenotype Correlations
5.2. Wolfram Syndrome Type 2 Genotype–Phenotype Correlations
6. Genetic Analysis of Wolfram Syndrome
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wolfram, D.J.; Wagener, H.P. Diabetes mellitus and simple optic atrophy among siblings: Report of four cases. Mayo. Clinproc. 1938, 13, 715–718. [Google Scholar]
- Rose, F.C.; Fraser, G.R.; Friedmann, A.I.; Kohner, E.M. The association of juvenile diabetes mellitus and optic atrophy: Clinical and genetical aspects. Quart. J. Med. 1966, 35, 385–405. [Google Scholar] [PubMed]
- Polymeropoulos, M.H.; Swift, R.G.; Swift, M. Linkage of the gene for Wolfram syndrome to markers on the short arm of chromosome 4. Nat. Genet. 1994, 8, 95–97. [Google Scholar] [CrossRef]
- Inoue, H.; Tanizawa, Y.; Wasson, J.; Behn, P.; Kalidas, K.; Bernal-Mizrachi, E.; Mueckler, M.; Marshall, H.; Donis-Keller, H.; Crock, P.; et al. A gene encoding a trans membrane protein is mutated in patients with diabetes mellitus and optic atrophy. Nat. Genet. 1998, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- El-Shanti, H.; Lidral, A.C.; Jarrah, N.; Druhan, L.; Ajlouni, K. Homozygosity mapping identifies an additional locus for Wolfram syndrome on chromosome 4q. Am. J. Hum. Genet. 2000, 66, 1229–1236. [Google Scholar] [CrossRef] [Green Version]
- Amr, S.; Heisey, C.; Zhang, M.; Xia, X.J.; Shows, K.H.; Ajlouni, K.; Pandya, A.; Satin, L.S.; El-Shanti, H.; Shiang, R.A. Homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 2007, 81, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.G.; Bundey, S.E.; Macleod, A.F. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995, 346, 1458–1463. [Google Scholar] [CrossRef]
- Matsunaga, K.; Tanabe, K.; Inoue, H.; Okuya, S.; Ohta, Y.; Akiyama, M.; Taguchi, A.; Kora, Y.; Okayama, N.; Yamada, Y.; et al. Wolfram syndrome in the Japanese population; molecular analysis of WFS1 gene and characterization of clinical features. PLoS ONE 2014, 9, e106906. [Google Scholar] [CrossRef] [Green Version]
- Zalloua, P.A.; Azar, S.T.; Delépine, M.; Makhoul, N.J.; Blanc, H.; Sanyoura, M.; Lavergne, A.; Stankov, K.; Lemainque, A.; Baz, P.; et al. WFS1 mutations are frequent monogenic causes of juvenile-onset diabetes mellitus in Lebanon. Hum. Mol. Genet. 2008, 17, 4012–4021. [Google Scholar] [CrossRef] [PubMed]
- Mozzillo, E.; Salzano, G.; Barbetti, F.; Maffeis, C.; Lombardo, F.; Franzese, A.; Delvecchio, M.; Marigliano, M. Survey on etiological diagnosis of diabetes in 1244 Italian diabetic children and adolescents: Impact of access to genetic testing. Diabetes Res. Clin. Pract. 2015, 107, e15–e18. [Google Scholar] [CrossRef]
- Delvecchio, M.; Mozzillo, E.; Salzano, G.; Iafusco, D.; Frontino, G.; Patera, P.I.; Rabbone, I.; Cherubini, V.; Grasso, V.; Tinto, N.; et al. Diabetes Study Group of the Italian Society of Pediatric Endocrinology and Diabetes (ISPED). Monogenic Diabetes Accounts for 6.3% of Cases Referred to 15 Italian Pediatric Diabetes Centers During 2007 to 2012. J. Clin. Endocrinol. Metab. 2017, 102, 1826–1834. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, S.; Xu, K.; Chen, Y.; Fu, Q.; Gu, Y.; Shi, Y.; Zhang, M.; Sun, M.; Chen, H.; et al. High Prevalence of a Monogenic Cause in Han Chinese Diagnosed with Type 1 Diabetes, Partly Driven by Nonsyndromic Recessive WFS1 Mutations. Diabetes 2020, 69, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Pacaud, D.; Schwandt, A.; de Beaufort, C.; Casteels, K.; Beltrand, J.; Birkebaek, N.H.; Campagnoli, M.; Bratina, N.; Limbert, C.; Mp O’Riordan, S.; et al. A description of clinician reported diagnosis of type 2 diabetes and other non-type 1 diabetes included in a large international multicentered pediatric diabetes registry (SWEET). Pediatr. Diabetes 2016, 17 (Suppl. 23), 24–31. [Google Scholar] [CrossRef] [Green Version]
- Cano, A.; Rouzier, C.; Monnot, S.; Chabrol, B.; Conrath, J.; Lecomte, P.; Delobel, B.; Boileau, P.; Valero, R.; Procaccio, V.; et al. Identification of novel mutations in WFS1 and genotype-phenotype correlation in Wolfram syndrome. Am. J. Med. Genet. A 2007, 143A, 1605–1612. [Google Scholar] [CrossRef]
- Tranebjærg, L.; Barrett, T.; Rendtorff, N.D. WFS1 Wolfram Syndrome Spectrum Disorder; 2009 February 24 [updated 2020 April 9]; GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2021. [Google Scholar] [PubMed]
- Pennings, R.J.; Huygen, P.L.; van den Ouweland, J.M.; Cryns, K.; Dikkeschei, L.D.; Van Camp, G.; Cremers, C.W. Sex-related hearing impairment in Wolfram syndrome patients identified by inactivating WFS1 mutations. Audiol. Neurootol. 2004, 9, 51–62. [Google Scholar] [CrossRef]
- Karzon, R.; Narayanan, A.; Chen, L.; Lieu, J.E.C.; Hershey, T. Longitudinal hearing loss in Wolfram syndrome. Orphanet. J. Rare Dis. 2018, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Ishigaki, S.; Oslowski, C.M.; Lu, S.; Lipson, K.L.; Ghosh, R.; Hayashi, E.; Ishihara, H.; Oka, Y.; Permutt, M.A.; et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Investig. 2010, 120, 744–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakdemirli, E.; Karabulut, N.; Bir, L.S.; Sermez, Y. Cranial magnetic resonance imaging of Wolfram (DIDMOAD) syndrome. Australas Radiol. 2005, 49, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Domenech, E.; Gomez-Zaera, M.; Nunes, V. Wolfram/DIDMOAD syndrome, a heterogenic and molecularly complex neurodegenerative disease. Pediatr. Endocrinol. Rev. 2006, 3, 249–257. [Google Scholar]
- Ito, S.; Sakakibara, R.; Hattori, T. Wolfram syndrome presenting marked brain MR imaging abnormalities with few neurologic abnormalities. AJNR Am. J. Neuroradiol. 2007, 28, 305–306. [Google Scholar] [PubMed]
- Swift, R.G.; Polymeropoulos, M.H.; Torres, R.; Swift, M. Predisposition of Wolfram syndrome heterozygotes to psychiatric illness. Mol. Psychiatry 1998, 3, 86–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Heredia, M.L.; Clèries, R.; Nunes, V. Genotypic classification of patients with Wolfram syndrome: Insights into the natural history of the disease and correlation with phenotype. Genet. Med. 2013, 15, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Rigoli, L.; Aloi, C.; Salina, A.; Di Bella, C.; Salzano, G.; Caruso, R.; Mazzon, E.; Maghnie, M.; Patti, G.; D’Annunzio, G.; et al. Wolfram syndrome 1 in the Italian population: Genotype-phenotype correlations. Pediatr. Res. 2020, 87, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Al-Sheyyab, M.; Jarrah, N.; Younis, E.; Shennak, M.M.; Hadidi, A.; Awidi, A.; El-Shanti, H.; Ajlouni, K. Bleeding tendency in Wolfram syndrome: A newly identified feature with phenotype genotype correlation. Eur. J. Pediatr. 2001, 160, 243–246. [Google Scholar] [CrossRef]
- Mozzillo, E.; Delvecchio, M.; Carella, M.; Grandone, E.; Palumbo, P.; Salina, A.; Aloi, C.; Buono, P.; Izzo, A.; D’Annunzio, G.; et al. A novel CISD2 intragenic deletion, optic neuropathy and platelet aggregation defect in Wolfram syndrome type 2. BMC Med. Genet. 2014, 15, 88. [Google Scholar] [CrossRef] [Green Version]
- Ajlouni, K.; Jarrah, N.; El-Khateeb, M.; El-Zaheri, M.; El Shanti, H.; Lidral, A. A Wolfram syndrome: Identification of a phenotypic and genotypic variant from Jordan. Am. J. Med. Genet. 2002, 115, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, M.T.; Tascini, G.; Crispoldi, R.; Orabona, C.; Mondanelli, G.; Grohmann, U.; Esposito, S. Wolfram syndrome, a rare neurodegenerative disease: From pathogenesis to future treatment perspectives. J. Transl. Med. 2019, 17, 238. [Google Scholar] [CrossRef] [Green Version]
- Urano, F. Wolfram syndrome: Diagnosis, management, and treatment. Curr. Diabetes Rep. 2016, 16, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci. Transl. Med. 2013, 5, 211ra156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadan, J.W.; Steiner, S.R.; O’Neill, C.M.; Nunemaker, C.S. The central role of calcium in the effects of cytokines on beta-cell function: Implications for type 1 and type 2 diabetes. Cell Calcium 2011, 50, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.; Hua, H.; Foo, K.; Martinez, H.; Watanabe, K.; Zimmer, M.; Kahler, D.J.; Freeby, M.; Chung, W.; LeDuc, C.; et al. beta-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 2014, 63, 923–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, T.; Gerbershagen, M.U.; Fiege, M.; Weisshorn, R.; Wappler, F. Dantrolene–a review of its pharmacology, therapeutic use and new developments. Anaesthesia 2004, 59, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Zatyka, M.; Da Silva Xavier, G.; Bellomo, E.A.; Leadbeater, W.; Astuti, D.; Smith, J.; Michelangeli, F.; Rutter, G.A.; Barrett, T.G. Sarco (endo) plasmic reticulum ATPase is a molecular partner of Wolfram syndrome 1 protein, which negatively regulates its expression. Hum. Mol. Genet. 2014, 24, 814–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, S.; Philbrook, C.; Gerbitz, K.D.; Bauer, M.F. Wolfram syndrome: Structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum. Mol. Genet. 2003, 12, 2003–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Inoue, H.; Tanizawa, Y.; Matsuzaki, Y.; Oba, J.; Watanabe, Y.; Shinoda, K.; Oka, Y. WFS1 (Wolfram syndrome 1) gene product: Predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum. Mol. Genet. 2001, 10, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.F.; Kao, C.H.; Chen, Y.T.; Wang, C.; Wu, C.; Tsai, C.; Liu, F.; Yang, C.; Wei, Y.; Hsu, M.; et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 15, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Rouzier, C.; Moore, D.; Delorme, C.; Lacas-Gervais, S.; Ait-El-Mkadem, S.; Fragaki, K.; Burté, F.; Serre, V.; Bannwarth, S.; Chaussenot, A.; et al. A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions. Hum. Mol. Gen. 2017, 26, 1599–1611. [Google Scholar] [CrossRef]
- Wang, C.-H.; Chen, Y.-F.; Wu, C.-Y.; Wu, P.-C.; Huang, Y.-L.; Kao, C.-H.; Lin, C.-H.; Kao, L.-S.; Tsai, T.-F.; Wei, Y.-H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2þ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-H.; Tsai, T.-F.; Wei, Y.-H. Role of mitochondrial dysfunction and dysregulation of Ca(2þ) homeostasis in insulin insensitivity of mammalian cells. Ann. N. Y. Acad. Sci. 2015, 1350, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Solovyova, N.; Veselovsky, N.; Toescu, E.C.; Verkhratsky, A. Ca(2+) dynamics in the lumen of the endoplasmic reticulum in sensory neurons: Direct visualization of Ca(2+)-induced Ca(2+) release triggered by physiological Ca(2+) entry. EMBO J. 2002, 21, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Wu, C.Y.; Kirby, R.; Kao, C.H.; Tsai, T.F. A role for the CISD2 gene in lifespan control and human disease. Ann. N. Y. Acad. Sci. 2010, 1201, 58–64. [Google Scholar] [CrossRef]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010, 29, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.E.; Andreyev, A.Y.; Divakaruni, A.S.; Karisch, R.; Perkins, G.; Wall, E.A.; van der Geer, P.; Chen, Y.-F.; Tsai, T.-F.; Simon, M.I.; et al. Wolfram Syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2þ homeostasis. EMBO Mol. Med. 2013, 5, 904–918. [Google Scholar] [CrossRef]
- Rigoli, L.; Bramanti, P.; Di Bella, C.; De Luca, F. Correction: Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr. Res. 2018, 84, 787, Erratum in Pediatr Res. 2018, 83, 921–929. [Google Scholar] [CrossRef]
- Chaussenot, A.; Rouzier, C.; Quere, M.; Plutino, M.; Ait-El-Mkadem, S.; Bannwarth, S.; Barth, M.; Dollfus, H.; Charles, P.; Nicolino, M.; et al. Mutation update and uncommon phenotypes in a French cohort of 96 patients with WFS1- related disorders. Clin. Genet. 2015, 87, 430–439. [Google Scholar] [CrossRef] [PubMed]
- d’Annunzio, G.; Minuto, N.; D’Amato, E.; de Toni, T.; Lombardo, F.; Pasquali, L.; Lorini, R. Wolfram syndrome (diabetes insipidus, diabetes, optic atrophy, and deafness): Clinical and genetic study. Diabetes Care 2008, 31, 1743–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astuti, D.; Sabir, A.; Fulton, P.; Zatyka, M.; Williams, D.; Hardy, C.; Milan, G.; Favaretto, F.; Yu-Wai-Man, P.; Rohayem, J.; et al. Monogenic diabetes syndromes: Locus-specific databases for Alström, Wolfram, and Thiamine-responsive megaloblastic anemia. Hum. Mutat 2017, 38, 764–777. [Google Scholar] [CrossRef]
- Eiberg, H.; Hansen, L.; Kjer, B.; Hansen, T.; Pedersen, O.; Bille, M.; Rosenberg, T.; Tranebjaerg, L. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J. Med. Genet. 2006, 43, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Berry, V.; Gregory-Evans, C.; Emmett, W.; Waseem, N.; Raby, J.; Prescott, D.; Moore, A.T.; Bhattacharya, S.S. Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur. J. Hum. Genet. 2013, 21, 1356–1360. [Google Scholar] [CrossRef] [Green Version]
- Bonnycastle, L.L.; Chines, P.S.; Hara, T.; Huyghe, J.R.; Swift, A.J.; Heikinheimo, P.; Mahadevan, J.; Peltonen, S.; Huopio, H.; Nuutila, P.; et al. Autosomal dominant diabetes arising from a Wolfram syndrome 1 mutation. Diabetes 2013, 62, 3943–3950. [Google Scholar] [CrossRef] [Green Version]
- Elli, F.M.; Ghirardello, S.; Giavoli, C.; Gangi, S.; Dioni, L.; Crippa, M.; Finelli, P.; Bergamaschi, S.; Mosca, F.; Spada, A.; et al. A new structural rearrangement associated to Wolfram syndrome in a child with a partial phenotype. Gene 2012, 509, 168–172. [Google Scholar] [CrossRef]
- Morikawa, S.; Tajima, T.; Nakamura, A.; Ishizu, K.; Ariga, T. A novel heterozygous mutation of the WFS1 gene leading to constitutive endoplasmic reticulum stress is the cause of Wolfram syndrome. Pediatr. Diabetes 2017, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- De Franco, E.; Flanagan, S.E.; Yagi, T.; Abreu, V.; Mahadevan, J.; Johnson, M.B.; Jones, G.; Acosta, F.; Mulaudzi, M.; Lek, N.; et al. Dominant ER stress-inducing WFS1 mutations underlie a genetic syndrome of neonatal/infancy-onset diabetes, congenital sensorineural deafness, and congenital cataracts. Diabetes 2017, 66, 2044–2053. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, D.T.; Manolakos, E.; Bothou, C.; Zoupanos, G.; Papoulidis, I.; Orru, S.; Skarmoutsos, F.; Delides, A.; Bakoula, C.; Papadimitriou, A.; et al. Maternal uniparental disomy of chromosome 4 and homozygous novel mutation in the WFS1 gene in a paediatric patient with Wolfram syndrome. Diabetes Metab. 2015, 41, 433–435. [Google Scholar] [CrossRef]
- Rendtorff, N.D.; Lodahl, M.; Boulahbel, H.; Johansen, I.R.; Pandya, A.; Welch, K.O.; Norris, V.W.; Arnos, K.S.; Bitner-Glindzicz, M.; Emery, S.B.; et al. Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. Am. J. Med. Genet. A 2011, 155A, 1298–1313. [Google Scholar] [CrossRef] [Green Version]
- Riachi, M.; Yilmaz, S.; Kurnaz, E.; Aycan, Z.; Çetinkaya, S.; Tranebjærg, L.; Rendtorff, N.D.; Bitner-Glindzicz, M.; Bockenhauer, D.; Hussain, K. Functional assessment of variants associated with Wolfram syndrome. Hum. Mol. Genet. 2019, 28, 3815–3824. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; La Sala, L.; Rondinelli, M.; Errichiello, E.; Zuffardi, O.; Puca, A.A.; Genovese, S.; Ceriello, A. A donor splice site mutation in CISD2 generates multiple truncated, non-functional isoforms in Wolfram syndrome type 2 patients. BMC Med. Genet. 2017, 18, 147. [Google Scholar] [CrossRef]
- Pourreza, M.R.; Sobhani, M.; Rahimi, A.; Aramideh, M.; Kajbafzadeh, A.M.; Noori-Daloii, M.R.; Tabatabaiefar, M.A. Homozygosity mapping and direct sequencing identify a novel pathogenic variant in the CISD2 gene in an Iranian Wolfram syndrome family. Acta Diabetol. 2020, 57, 81–87. [Google Scholar] [CrossRef]
- Rondinelli, M.; Novara, F.; Calcaterra, V.; Zuffardi, O.; Genovese, S. Wolfram syndrome 2: A novel CISD2 mutation identified in Italian siblings. Acta Diabetol. 2015, 52, 175–178. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Major Clinical Findings | Other Clinical Findings |
|---|---|
| Diabetes mellitus (a) Age at diagnosis: 6–10 years | Urinary tract problems and renal dysfunction (neurogenic bladder, bladder incontinence, urinary tract infection) Age of diagnosis: second decade of life |
| Optic atrophy (a) Age at diagnosis: 10–15 years | Psychiatric symptoms (depression, psychosis, panic attacks, sleep abnormalities, mood swings) |
| Diabetes insipidus Age at diagnosis: 14–20 years | Neurological manifestation/autonomic dysfunction (central apnea, dysphagia, areflexia, epilepsy, decreased ability to taste and detect odors, headache, orthostatic hypotension, hypothermia, hyperpyrexia, gastroparesis, constipation) |
| Sensorineural hearing loss Age at diagnosis: 16–20 years | Endocrine disorders (hypogonadism, growth hormone deficiency, corticotropin deficiency, delayed menarche) |
| Ataxia Age at diagnosis: 15–25 years | Dominant disease with or without diabetes mellitus and recessive Wolfram-like disease without diabetes mellitus |
| Upper intestinal ulcers and platelet aggregation defect (b) |
| Groups | Localization | Type | Alterations |
|---|---|---|---|
| 1 | before exon 8 | nonsense and frameshift | complete deletion |
| 2 | aa 1–670 aa 701–890 | missense nonsense | complete degradation |
| 3 | after exon 8 and before aa700 after exon 8 aa 671–700 | nonsense frameshift missense | defective or trun-cated protein |
| Class | Functional Alterations |
|---|---|
| A1 | Wolframin depletion due to WFS1 mRNA degradation |
| A2 | Wolframin depletion due to WFS1 mRNA and protein degradation |
| A3 | Wolframin depletion due to protein degradation |
| B | Reduced expression of defective Wolframin |
| C | Expression of defective Wolframin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delvecchio, M.; Iacoviello, M.; Pantaleo, A.; Resta, N. Clinical Spectrum Associated with Wolfram Syndrome Type 1 and Type 2: A Review on Genotype–Phenotype Correlations. Int. J. Environ. Res. Public Health 2021, 18, 4796. https://doi.org/10.3390/ijerph18094796
Delvecchio M, Iacoviello M, Pantaleo A, Resta N. Clinical Spectrum Associated with Wolfram Syndrome Type 1 and Type 2: A Review on Genotype–Phenotype Correlations. International Journal of Environmental Research and Public Health. 2021; 18(9):4796. https://doi.org/10.3390/ijerph18094796
Chicago/Turabian StyleDelvecchio, Maurizio, Matteo Iacoviello, Antonino Pantaleo, and Nicoletta Resta. 2021. "Clinical Spectrum Associated with Wolfram Syndrome Type 1 and Type 2: A Review on Genotype–Phenotype Correlations" International Journal of Environmental Research and Public Health 18, no. 9: 4796. https://doi.org/10.3390/ijerph18094796
APA StyleDelvecchio, M., Iacoviello, M., Pantaleo, A., & Resta, N. (2021). Clinical Spectrum Associated with Wolfram Syndrome Type 1 and Type 2: A Review on Genotype–Phenotype Correlations. International Journal of Environmental Research and Public Health, 18(9), 4796. https://doi.org/10.3390/ijerph18094796