
Prenylation Defects and Oxidative Stress Trigger the Main Consequences of Neuroinflammation Linked to Mevalonate Pathway Deregulation

 ,
,  ,
, 
 ,
, 
 and
and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Mevalonate Pathway
3. Critical Points to Regulate the Metabolic Pathway of Cholesterol
4. The Prenylation Process
5. Consequences Caused by Prenylation Defects
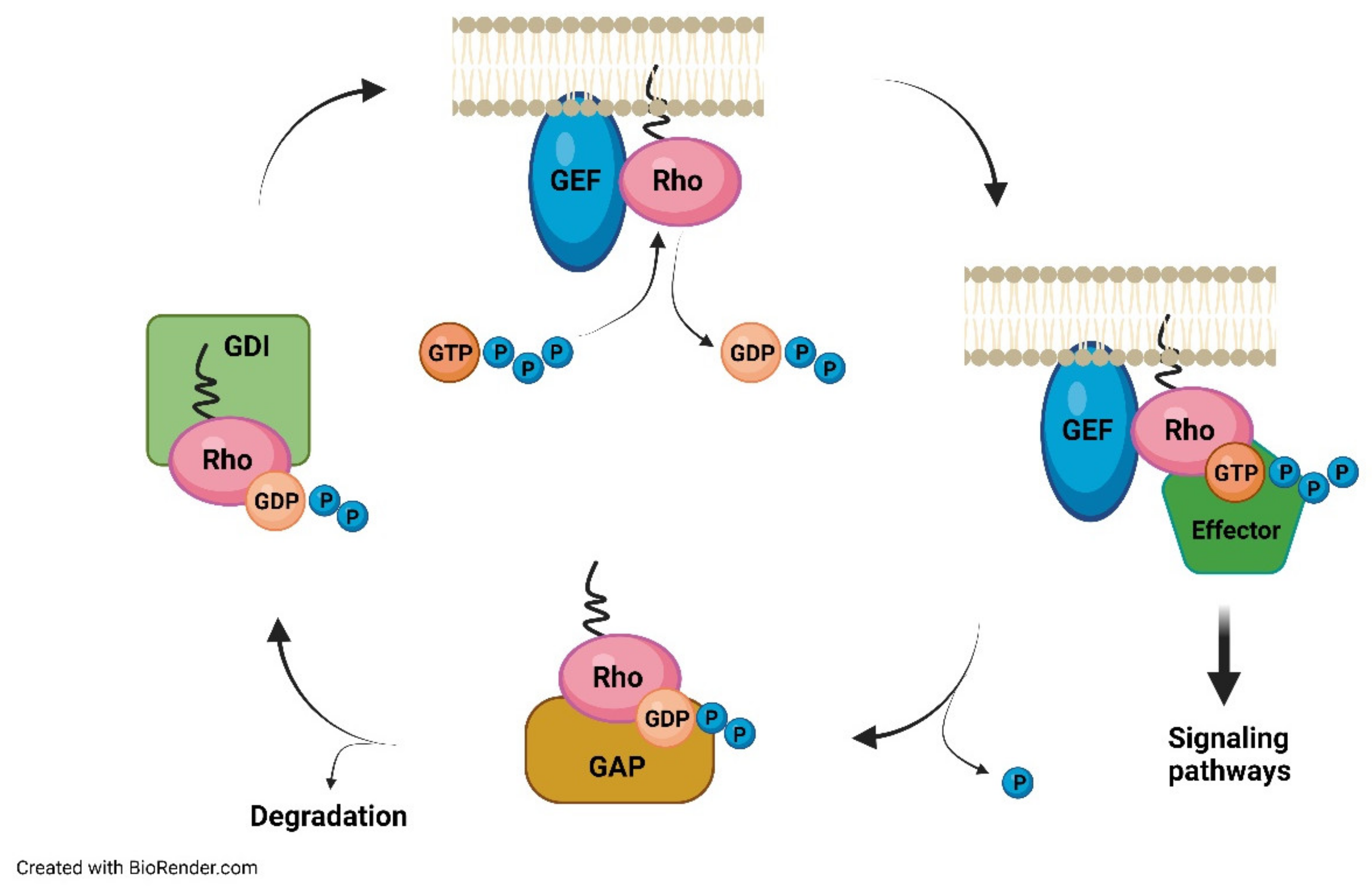
6. Neuroinflammation, Oxidative Stress, and Fever as a Consequence of Altered Mevalonate Pathway Flux
7. Coenzyme Q10: The Fine Regulation of Its Antioxidant Properties
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeong, A.; Suazo, K.F.; Wood, W.G.; Distefano, M.K.; Li, L. Isoprenoids and Protein Prenylation: Implications in the Pathogenesis and Therapeutic Intervention of Alzheimer’s Disease. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 279–310. [Google Scholar] [CrossRef] [PubMed]
- Desmet, S.; Morreel, K.; Dauwe, R. Origin and Function of Structural Diversity in the Plant Specialized Metabolome. Plants 2021, 10, 2393. [Google Scholar] [CrossRef] [PubMed]
- Lamphear, C.L.; Zverina, E.A.; Hougland, J.L.; Fierke, C.A. Global Identification of Protein Prenyltransferase Substrates: Defining the Prenylated Proteome. Enzymes 2011, 29, 207–234. [Google Scholar]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D. Phenotype variability of autoinflammatory disorders in the pediatric patient: A pictorial overview. J. Evid.-Based Med. 2020, 13, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Kawamukai, M. Biosynthesis and applications of prenylquinones. Biosci. Biotechnol. Biochem. 2018, 82, 963–977. [Google Scholar] [CrossRef]
- Rivett, E.D.; Heo, L.; Feig, M.; Hegg, E.L. Biosynthesis and trafficking of heme o and heme a: New structural insights and their implications for reaction mechanisms and prenylated heme transfer. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 640–668. [Google Scholar] [CrossRef]
- Pierrel, F.; Burgardt, A.; Lee, J.H.; Pelosi, L.; Wendisch, V.F. Recent advances in the metabolic pathways and microbial production of coenzyme Q. World J. Microbiol. Biotechnol. 2022, 38, 58. [Google Scholar] [CrossRef]
- Ciaglia, E.; Laezza, C.; Abate, M.; Pisanti, S.; Ranieri, R.; D’alessandro, A.; Picardi, P.; Gazzerro, P.; Bifulco, M. Recognition by natural killer cells of 6-isopentenyladenosine-treated human glioma cell lines. Int. J. Cancer 2018, 142, 176–190. [Google Scholar] [CrossRef] [Green Version]
- Mitsche, M.A.; McDonald, J.G.; Hobbs, H.H.; Cohen, J.C. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. eLife 2015, 4, e07999. [Google Scholar] [CrossRef] [PubMed]
- Tchernof, A.; Després, J.P. Pathophysiology of human visceral obesity: An update. Physiol. Rev. 2013, 93, 359–404. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L.; Westerterp, M.; Murphy, A.J. Cholesterol efflux: A novel regulator of myelopoiesis and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2547–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yvan-Charvet, L.; Bonacina, F.; Guinamard, R.R.; Norata, G.D. Immunometabolic function of cholesterol in cardiovascular disease and beyond. Cardiovasc. Res. 2019, 115, 1393–1407. [Google Scholar] [CrossRef] [PubMed]
- Lütjohann, D.; Björkhem, I.; Diczfalusy, U.; Ståhle, L.; Ahlborg, G.; Wahren, J. Cholesterol homeostasis in human brain: Turnover of 24(S)-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 1998, 39, 1594–1600. [Google Scholar]
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 2001, 12, 105–112. [Google Scholar] [CrossRef]
- Schumacher, M.M.; DeBose-Boyd, R.A. Posttranslational Regulation of HMG CoA Reductase, the Rate-Limiting Enzyme in Synthesis of Cholesterol. Annu. Rev. Biochem. 2021, 90, 659–679. [Google Scholar] [CrossRef]
- Pierzchlińska, A.; Droździk, M.; Białecka, M. A Possible Role for HMG-CoA Reductase Inhibitors and Its Association with HMGCR Genetic Variation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 12198. [Google Scholar] [CrossRef]
- Kwon, S.J.; Hong, K.W.; Choi, S.; Hong, J.S.; Kim, J.W.; Kim, J.W.; Lee, H.J.; Jang, H.B.; Yum, K.S. Association of 3-hydroxy-3-methylglutaryl-coenzyme A reductase gene polymorphism with obesity and lipid metabolism in children and adolescents with autism spectrum disorder. Metab. Brain Dis. 2022, 37, 319–328. [Google Scholar] [CrossRef]
- Wall, C.T.J.; Lefebvre, G.; Metairon, S.; Descombes, P.; Wiederkehr, A.; Santo-Domingo, J. Mitochondrial respiratory chain dysfunction alters ER sterol sensing and mevalonate pathway activity. J. Biol. Chem. 2022, 298, 101652. [Google Scholar] [CrossRef]
- Zhang, S. Natural history of mevalonate kinase deficiency: A literature review. Pediatr. Rheumatol. Online J. 2016, 14, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.; Qi, H.; Zhang, H.; Ding, L.; Huang, Q.; Zhao, D.; Wu, B.J.; Li, X. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front. Oncol. 2020, 10, 1510. [Google Scholar] [CrossRef] [PubMed]
- Hinson, D.D.; Chambliss, K.L.; Toth, M.J.; Tanaka, R.D.; Gibson, K.M. Post-translational regulation of mevalonate kinase by intermediates of the cholesterol and nonsterol isoprene biosynthetic pathways. J. Lipid Res. 1997, 38, 2216–2223. [Google Scholar] [CrossRef]
- Waku, T.; Hagiwara, T.; Tamura, N.; Atsumi, Y.; Urano, Y.; Suzuki, M.; Iwami, T.; Sato, K.; Yamamoto, M.; Noguchi, N.; et al. NRF3 upregulates gene expression in SREBP2-dependent mevalonate pathway with cholesterol uptake and lipogenesis inhibition. iScience 2021, 24, 103180. [Google Scholar] [CrossRef] [PubMed]
- Elhani, I.; Hentgen, V.; Grateau, G.; Georgin-Lavialle, S. Neurological manifestations in mevalonate kinase deficiency: A systematic review. Mol. Genet. Metab. 2022, 136, 85–93. [Google Scholar] [CrossRef]
- Caligiuri, G.; Norata, G.D. Fuel for thought: Immunometabolism is a paradigm shift in understanding immunity in cardiovascular disease. Cardiovasc. Res. 2019, 115, 1383–1384. [Google Scholar] [CrossRef]
- Nowaczyk, M.J.; Irons, M.B. Smith-Lemli-Opitz syndrome: Phenotype, natural history, and epidemiology. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 250–262. [Google Scholar] [CrossRef]
- Maresca, G.; Formica, C.; Nocito, V.; Latella, D.; Leonardi, S.; De Cola, M.C.; Triglia, G.; Bramanti, P.; Corallo, F. Neuropsychological assessment in Niemann-Pick disease type C: A systematic review. Neurol. Sci. 2021, 42, 3167–3175. [Google Scholar] [CrossRef]
- Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of Human Coenzyme Q10 Metabolism: An Overview. Int. J. Mol. Sci. 2020, 21, 6695. [Google Scholar] [CrossRef]
- Quinzii, C.M.; DiMauro, S.; Hirano, M. Human coenzyme Q10 deficiency. Neurochem. Res. 2007, 32, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Manzar, H.; Abdulhussein, D.; Yap, T.E.; Cordeiro, M.F. Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain. Int. J. Mol. Sci. 2020, 21, 9299. [Google Scholar] [CrossRef] [PubMed]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.B.; Korade, Z.; Mirnics, K. Cholesterol Biosynthesis and Uptake in Developing Neurons. ACS Chem. Neurosci. 2019, 10, 3671–3681. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Dis. Model. Mech. 2012, 5, 746–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, D.; Kelley, R.I.; Hoffmann, G.F. Inherited disorders of cholesterol biosynthesis. Neuropediatrics 2001, 32, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.L.; Casey, P.J. Protein prenylation: Molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996, 65, 241–269. [Google Scholar] [CrossRef] [PubMed]
- Casey, P.J.; Seabra, M.C. Protein Prenyltransferases. J. Biol. Chem. 1996, 71, 5289–5292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, Y.; Stradley, S.J.; Gierasch, L.M.; Brown, M.S.; Goldstein, J.L. Sequence Requirement for Peptide Recognition by the Rat Brain p21 Protein Farnesyltransferase. Proc. Natl. Acad. Sci. USA 1991, 88, 732–736. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, K.; Goodwin, G.W.; Ghomashchi, F.; Glomset, J.A.; Gelb, M.H. A protein geranylgeranyltransferase from bovine brain: Implications for protein prenylation specificity. Proc. Natl. Acad. Sci. USA 1991, 88, 5302–5306. [Google Scholar] [CrossRef] [Green Version]
- Blanden, M.J.; Suazo, K.F.; Hildebrandt, E.R.; Hardgrove, D.S.; Patel, M.; Saunders, W.P.; Distefano, M.D.; Schmidt, W.K.; Hougland, J.L. Efficient farnesylation of an extended C-terminal C(x)3X sequence motif expands the scope of the prenylated proteome. J. Biol. Chem. 2018, 293, 2770–2785. [Google Scholar] [CrossRef] [Green Version]
- Wiemer, A.J.; Wiemer, D.F.; Hohl, R.J. Geranylgeranyl diphosphate synthase: An emerging therapeutic target. Clin. Pharmacol. Ther. 2011, 90, 804–812. [Google Scholar] [CrossRef]
- Noguera-Salvà, M.A.; Guardiola-Serrano, F.; Martin, M.L.; Marcilla-Etxenike, A.; Bergo, M.O.; Busquets, X.; Escribá, P.V. Role of the C-terminal basic amino acids and the lipid anchor of the Gγ2 protein in membrane interactions and cell localization. Biochim. Biophys. Acta Biomembr. 2017, 859, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Hampton, S.E.; Dore, T.M.; Schmidt, W.K. Rce1: Mechanism and inhibition. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Borini Etichetti, C.M.; Arel Zalazar, E.; Cocordano, N.; Girardini, J. Beyond the Mevalonate Pathway: Control of Post-Prenylation Processing by Mutant p53. Front. Oncol. 2020, 10, 595034. [Google Scholar] [CrossRef]
- Jun, J.E.; Rubio, I.; Roose, J.P. Regulation of Ras Exchange Factors and cellular localization of Ras activation by lipid messengers in T cells. Front. Immunol. 2013, 4, 239. [Google Scholar] [CrossRef] [Green Version]
- Bifulco, M. Role of the isoprenoid pathway in Ras transforming activity, cytoskeleton organization, cell proliferation and apoptosis. Life Sci. 2005, 77, 1740–1749. [Google Scholar] [CrossRef]
- Prior, I.A.; Hancock, J.F. Compartmentalization of Ras proteins. J. Cell Sci. 2001, 114, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Lane, K.T.; Beese, L.S. Thematic review series: Lipid posttranslational modifications. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J. Lipid Res. 2006, 47, 681–699. [Google Scholar] [CrossRef] [Green Version]
- Sindhu, M.; Saini, V.; Piplani, S.; Kumar, A. Molecular Dynamics of Rab7::REP1::GGTase-II Ternary Complex and Identification of Their Putative Drug Binding Sites. Indian J. Pharm. Sci. 2013, 75, 23–30. [Google Scholar]
- Marchwicka, A.; Kamińska, D.; Monirialamdari, M.; Błażewska, K.M.; Gendaszewska-Darmach, E. Protein Prenyltransferases and Their Inhibitors: Structural and Functional Characterization. Int. J. Mol. Sci. 2022, 23, 5424. [Google Scholar] [CrossRef]
- Watterson, A.; Tatge, L.; Wajahat, N.; Arneaud, S.L.B.; Solano Fonseca, R.; Beheshti, S.T.; Metang, P.; Mihelakis, M.; Zuurbier, K.R.; Corley, C.D.; et al. Intracellular lipid surveillance by small G protein geranylgeranylation. Nature 2022, 605, 736–740. [Google Scholar] [CrossRef]
- Manaswiyoungkul, P.; de Araujo, E.D.; Gunning, P.T. Targeting prenylation inhibition through the mevalonate pathway. RSC Med. Chem. 2019, 11, 51–71. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, T.-Y.; Zhao, M.-F.; Li, C.-J. The balance of protein farnesylation and geranylgeranylation during the progression of nonalcoholic fatty liver disease. J. Biol. Chem. 2020, 295, 5152–5162. [Google Scholar] [CrossRef] [Green Version]
- Waller, D.D.; Park, J.; Tsantrizos, Y.S. Inhibition of farnesyl pyrophosphate (FPP) and/or geranylgeranyl pyrophosphate (GGPP) biosynthesis and its implication in the treatment of cancers. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 41–60. [Google Scholar] [CrossRef]
- Laezza, C.; Notarnicola, M.; Caruso, M.G.; Messa, C.; Macchia, M.; Bertini, S.; Minutolo, F.; Portella, G.; Fiorentino, L.; Stingo, S.; et al. N6-isopentenyladenosine arrests tumor cell proliferation by inhibiting farnesyl diphosphate synthase and protein prenylation. FASEB J. 2006, 20, 412–418. [Google Scholar] [CrossRef]
- Roy, M.; Kung, H.J.; Ghosh, P.M. Statins and prostate cancer: Role of cholesterol inhibition vs. prevention of small GTP-binding proteins. Am. J. Cancer Res. 2011, 1, 542–561. [Google Scholar]
- Todenhöfer, T.; Hennenlotter, J.; Kühs, U.; Gerber, V.; Gakis, G.; Vogel, U.; Aufderklamm, S.; Merseburger, A.; Knapp, J.; Stenzl, A.; et al. Altered expression of farnesyl pyrophosphate synthase in prostate cancer: Evidence for a role of the mevalonate pathway in disease progression? World J. Urol. 2013, 31, 345–350. [Google Scholar] [CrossRef]
- Reddy, J.M.; Raut, N.G.R.; Seifert, J.L.; Hynds, D.L. Regulation of Small GTPase Prenylation in the Nervous System. Mol. Neurobiol. 2020, 57, 2220–2231. [Google Scholar] [CrossRef]
- Hottman, D.; Li, L. Protein prenylation and synaptic plasticity: Implications for Alzheimer’s disease. Mol. Neurobiol. 2014, 50, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Selzer, M.E. RhoA as a target to promote neuronal survival and axon regeneration. Neural Regen. Res. 2017, 12, 525–528. [Google Scholar]
- Munoz, M.A.; Jurczyluk, J.; Simon, A.; Hissaria, P.; Arts, R.J.W.; Coman, D.; Boros, C.; Mehr, S.; Rogers, M.J. Defective Protein Prenylation in a Spectrum of Patients with Mevalonate Kinase Deficiency. Front. Immunol. 2019, 10, 1900. [Google Scholar] [CrossRef] [Green Version]
- Jeyaratnam, J.; Frenkel, J. Management of Mevalonate Kinase Deficiency: A Pediatric Perspective. Front. Immunol. 2020, 11, 1150. [Google Scholar] [CrossRef]
- Politiek, F.A.; Waterham, H.R. Compromised Protein Prenylation as Pathogenic Mechanism in Mevalonate Kinase Deficiency. Front. Immunol. 2021, 12, 724991. [Google Scholar] [CrossRef]
- Munoz, M.A.; Jurczyluk, J.; Mehr, S.; Chai, R.C.; Arts, R.J.; Sheu, A.; McMahon, C.; Center, J.R.; Singh-Grewal, D.; Chaitow, J.; et al. Defective protein prenylation is a diagnostic biomarker of mevalonate kinase deficiency. J. Allergy Clin. Immunol. 2017, 140, 873–875.e6. [Google Scholar] [CrossRef] [Green Version]
- Jurczyluk, J.; Munoz, M.A.; Skinner, O.P.; Chai, R.C.; Ali, N.; Palendira, U.; Quinn, J.M.; Preston, A.; Tangye, S.G.; Brown, A.J.; et al. Mevalonate kinase deficiency leads to decreased prenylation of Rab GTPases. Immunol. Cell Biol. 2016, 94, 994–999. [Google Scholar] [CrossRef] [Green Version]
- Skinner, O.P.; Jurczyluk, J.; Baker, P.J.; Masters, S.L.; Rios Wilks, A.G.; Clearwater, M.S.; Robertson, A.A.B.; Schroder, K.; Mehr, S.; Munoz, M.A.; et al. Lack of protein prenylation promotes NLRP3 inflammasome assembly in human monocytes. J. Allergy Clin. Immunol. 2019, 143, 2315–2317. [Google Scholar] [CrossRef] [Green Version]
- van der Burgh, R.; Pervolaraki, K.; Turkenburg, M.; Waterham, H.R.; Frenkel, J.; Boes, M. Unprenylated RhoA contributes to IL-1β hypersecretion in mevalonate kinase deficiency model through stimulation of Rac1 activity. J. Biol. Chem. 2014, 289, 27757–27765. [Google Scholar] [CrossRef] [Green Version]
- Favier, L.A.; Schulert, G.S. Mevalonate kinase deficiency: Current perspectives. Appl. Clin. Genet. 2016, 9, 101–110. [Google Scholar]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dai, L.; Deyuan, L. Mitophagy in neurological disorders. J. Neuroinflamm. 2021, 18, 297. [Google Scholar] [CrossRef]
- Kaur, N.; Chugh, H.; Sakharkar, M.K.; Dhawan, U.; Chidambaram, S.B.; Chandra, R. Neuroinflammation Mechanisms and Phytotherapeutic Intervention: A Systematic Review. ACS Chem. Neurosci. 2020, 11, 3707–3731. [Google Scholar] [CrossRef]
- Stolp, H.B.; Dziegielewska, K.M. Role of developmental inflammation and blood-brain barrier dysfunction in neurodevelopmental and neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 2009, 35, 132–146. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Wu, L.J. How microglia sense and regulate neuronal activity. Glia 2021, 69, 1637–1653. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and Astrocytes in Disease: Dynamic Duo or Partners in Crime? Trends Immunol. 2020, 41, 820–835. [Google Scholar] [CrossRef]
- Rodríguez, A.M.; Rodríguez, J.; Giambartolomei, G.H. Microglia at the Crossroads of Pathogen-Induced Neuroinflammation. ASN Neuro 2022, 14, 17590914221104566. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Borst, K.; Schwabenland, M.; Prinz, M. Microglia metabolism in health and disease. Neurochem. Int. 2019, 130, 104331. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Uccelli, A.; Gattorno, M. Neurological manifestations in autoinflammatory diseases. Clin. Exp. Rheumatol. 2018, 36, 61–67. [Google Scholar]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- Hasday, J.D.; Singh, I.S. Fever and the heat shock response: Distinct, partially overlapping processes. Cell Stress Chaperones 2000, 5, 471–480. [Google Scholar] [CrossRef]
- Blatteis, C.M.; Li, S.; Li, Z.; Feleder, C.; Perlik, V. Cytokines, PGE2 and endotoxic fever: A re-assessment. Prostaglandins Other Lipid Mediat. 2005, 76, 1–18. [Google Scholar] [CrossRef]
- Garami, A.; Steiner, A.A.; Romanovsky, A.A. Fever and hypothermia in systemic inflammation. Handb. Clin. Neurol. 2018, 157, 565–597. [Google Scholar]
- Szarka, A.; Lőrincz, T.; Hajdinák, P. Friend or Foe: The Relativity of (Anti)oxidative Agents and Pathways. Int. J. Mol. Sci. 2022, 23, 5188. [Google Scholar] [CrossRef]
- Patergnani, S.; Morciano, G.; Carinci, M.; Leo, S.; Pinton, P.; Rimessi, A. The “mitochondrial stress responses”: The “Dr. Jekyll and Mr. Hyde” of neuronal disorders. Neural Regen. Res. 2022, 17, 2563–2575. [Google Scholar]
- Vitória, J.J.M.; Trigo, D.; da Cruz, E.; Silva, O.A.B. Revisiting APP secretases: An overview on the holistic effects of retinoic acid receptor stimulation in APP processing. Cell. Mol. Life Sci. 2022, 79, 101. [Google Scholar] [CrossRef]
- Zindler, E.; Zipp, F. Neuronal injury in chronic CNS inflammation. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 551–562. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Kleiner, G.; Valencic, E.; Campisciano, G.; Girardelli, M.; Crovella, S.; Knowles, A.; Marcuzzi, A. Block of the mevalonate pathway triggers oxidative and inflammatory molecular mechanisms modulated by exogenous isoprenoid compounds. Int. J. Mol. Sci. 2014, 15, 6843–6856. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Vince, J.E. Ambiguities in NLRP3 inflammasome regulation: Is there a role for mitochondria? Biochim. Biophys. Acta 2014, 1840, 1433–1440. [Google Scholar] [CrossRef]
- Yin, Y.; Yan, Y.; Jiang, X.; Mai, J.; Chen, N.C.; Wang, H.; Yang, X.-F. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int. J. Immunopathol. Pharmacol. 2009, 22, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Santos-Ocaña, C.; Cascajo, M.V.; Alcázar-Fabra, M.; Staiano, C.; López-Lluch, G.; Brea-Calvo, G.; Navas, P. Cellular Models for Primary CoQ Deficiency Pathogenesis Study. Int. J. Mol. Sci. 2021, 22, 10211. [Google Scholar] [CrossRef]
- Akula, M.K.; Shi, M.; Jiang, Z.; Foster, C.E.; Miao, D.; Li, A.S.; Zhang, X.; Gavin, R.M.; Forde, S.D.; Germain, G.; et al. Control of the innate immune response by the mevalonate pathway. Nat. Immunol. 2016, 17, 922–929. [Google Scholar] [CrossRef]
- Masumoto, J.; Zhou, W.; Morikawa, S.; Hosokawa, S.; Taguchi, H.; Yamamoto, T.; Kurata, M.; Kaneko, N. Molecular biology of autoinflammatory diseases. Inflamm. Regen. 2021, 41, 33. [Google Scholar] [CrossRef]
- Okin, D.; Medzhitov, R. The Effect of Sustained Inflammation on Hepatic Mevalonate Pathway Results in Hyperglycemia. Cell 2016, 165, 343–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, G.P.; Hooff, G.P.; Strandjord, D.M.; Igbavboa, U.; Volmer, D.A.; Müller, W.E.; Wood, W.G. Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiol. Dis. 2009, 35, 251–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonini, C.; Segatto, M.; Pallottini, V. Impact of Sex and Age on the Mevalonate Pathway in the Brain: A Focus on Effects Induced by Maternal Exposure to Exogenous Compounds. Metabolites 2020, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, X.; Li, L.; Ma, X.; Yang, C.; Liu, Z.; Li, C.; Fernandez-Cabezudo, M.J.; Al-Ramadi, B.K.; Wu, C.; et al. Farnesyl pyrophosphate is a new danger signal inducing acute cell death. PLoS Biol. 2021, 19, e3001134. [Google Scholar] [CrossRef]
- Marcuzzi, A.; Zanin, V.; Piscianz, E.; Tricarico, P.M.; Vuch, J.; Girardelli, M.; Monasta, L.; Bianco, A.M.; Crovella, S. Lovastatin-induced apoptosis is modulated by geranylgeraniol in a neuroblastoma cell line. Int. J. Dev. Neurosci. 2012, 30, 451–456. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Marcuzzi, A.; Piscianz, E.; Monasta, L.; Crovella, S.; Kleiner, G. Mevalonate kinase deficiency and neuroinflammation: Balance between apoptosis and pyroptosis. Int. J. Mol. Sci. 2013, 14, 23274–23288. [Google Scholar] [CrossRef] [Green Version]
- De Benedetti, F.; Gattorno, M.; Anton, J.; Ben-Chetrit, E.; Frenkel, J.; Hoffman, H.M.; Koné-Paut, I.; Lachmann, H.; Ozen, S.; Simon, A.; et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N. Engl. J. Med. 2018, 378, 1908–1919. [Google Scholar] [CrossRef] [Green Version]
- Grom, A.A.; Horne, A.; De Benedetti, F. Macrophage activation syndrome in the era of biologic therapy. Nat. Rev. Rheumatol. 2016, 12, 259–268. [Google Scholar] [CrossRef]
- Szkopińska, A. Ubiquinone. Biosynthesis of quinone ring and its isoprenoid side chain. Intracellular localization. Acta Biochim. Pol. 2000, 47, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Llanos-González, E.; Alcain, F.J. The Use of Coenzyme Q10 in Cardiovascular Diseases. Antioxidants 2021, 10, 755. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; de la Mata, M.; Villanueva-Paz, M.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Munuera, M.; et al. Atherosclerosis and Coenzyme Q10. Int. J. Mol. Sci. 2019, 20, 5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelzer, C.; Niklowitz, P.; Okun, J.G.; Haas, D.; Menke, T.; Döring, F. Ubiquinol-induced gene expression signatures are translated into altered parameters of erythropoiesis and reduced low density lipoprotein cholesterol levels in humans. IUBMB Life 2011, 63, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Somagutta, M.K.R.; Shama, N.; Pormento, M.K.L.; Jagani, R.P.; Ngardig, N.N.; Ghazarian, K.; Mahmutaj, G.; El-Faramawy, K.; Mahadevaiah, A.; Jain, M.S. Statin-induced necrotizing autoimmune myopathy: A systematic review. Reumatologia 2022, 60, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Sehgal, V.S.; Kashfi, K. Molecular targets of statins and their potential side effects: Not all the glitter is gold. Eur. J. Pharmacol. 2022, 922, 174906. [Google Scholar] [CrossRef]
- Aaseth, J.; Alexander, J.; Alehagen, U. Coenzyme Q10 supplementation—In ageing and disease. Mech. Ageing Dev. 2021, 197, 111521. [Google Scholar] [CrossRef]
- Raizner, A.E.; Quiñones, M.A. Coenzyme Q10 for Patients with Cardiovascular Disease: JACC Focus Seminar. J. Am. Coll. Cardiol. 2021, 77, 609–619. [Google Scholar] [CrossRef]
- Alcázar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar]
- Tan, W.; Airik, R. Primary coenzyme Q10 nephropathy, a potentially treatable form of steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 2021, 36, 3515–3527. [Google Scholar] [CrossRef]
- Schijvens, A.M.; van de Kar, N.C.; Bootsma-Robroeks, C.; Cornelissen, E.A.; van den Heuvel, L.P.; Schreuder, M.F. Mitochondrial Disease and the Kidney with a Special Focus o.on CoQ10 Deficiency. Kidney Int. Rep. 2020, 5, 2146–2159. [Google Scholar] [CrossRef]
- Caglayan, A.O.; Gumus, H.; Sandford, E.; Kubisiak, T.L.; Ma, Q.; Ozel, A.B.; Per, H.; Li, J.Z.; Shakkottai, V.G.; Burmeister, M. COQ4 Mutation Leads to Childhood-Onset Ataxia Improved by CoQ10 Administration. Cerebellum 2019, 18, 665–669. [Google Scholar] [CrossRef]
- Stefely, J.A.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.D.; et al. Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol. Cell 2016, 63, 608–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baschiera, E.; Sorrentino, U.; Calderan, C.; Desbats, M.A.; Salviati, L. The multiple roles of coenzyme Q in cellular homeostasis and their relevance for the pathogenesis of coenzyme Q deficiency. Free Radic. Biol. Med. 2021, 166, 277–286. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisanti, S.; Rimondi, E.; Pozza, E.; Melloni, E.; Zauli, E.; Bifulco, M.; Martinelli, R.; Marcuzzi, A. Prenylation Defects and Oxidative Stress Trigger the Main Consequences of Neuroinflammation Linked to Mevalonate Pathway Deregulation. Int. J. Environ. Res. Public Health 2022, 19, 9061. https://doi.org/10.3390/ijerph19159061
Pisanti S, Rimondi E, Pozza E, Melloni E, Zauli E, Bifulco M, Martinelli R, Marcuzzi A. Prenylation Defects and Oxidative Stress Trigger the Main Consequences of Neuroinflammation Linked to Mevalonate Pathway Deregulation. International Journal of Environmental Research and Public Health. 2022; 19(15):9061. https://doi.org/10.3390/ijerph19159061
Chicago/Turabian StylePisanti, Simona, Erika Rimondi, Elena Pozza, Elisabetta Melloni, Enrico Zauli, Maurizio Bifulco, Rosanna Martinelli, and Annalisa Marcuzzi. 2022. "Prenylation Defects and Oxidative Stress Trigger the Main Consequences of Neuroinflammation Linked to Mevalonate Pathway Deregulation" International Journal of Environmental Research and Public Health 19, no. 15: 9061. https://doi.org/10.3390/ijerph19159061
APA StylePisanti, S., Rimondi, E., Pozza, E., Melloni, E., Zauli, E., Bifulco, M., Martinelli, R., & Marcuzzi, A. (2022). Prenylation Defects and Oxidative Stress Trigger the Main Consequences of Neuroinflammation Linked to Mevalonate Pathway Deregulation. International Journal of Environmental Research and Public Health, 19(15), 9061. https://doi.org/10.3390/ijerph19159061