



Pan-Genome-Assisted Computational Design of a Multi-Epitopes-Based Vaccine Candidate against Helicobacter cinaedi

 , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
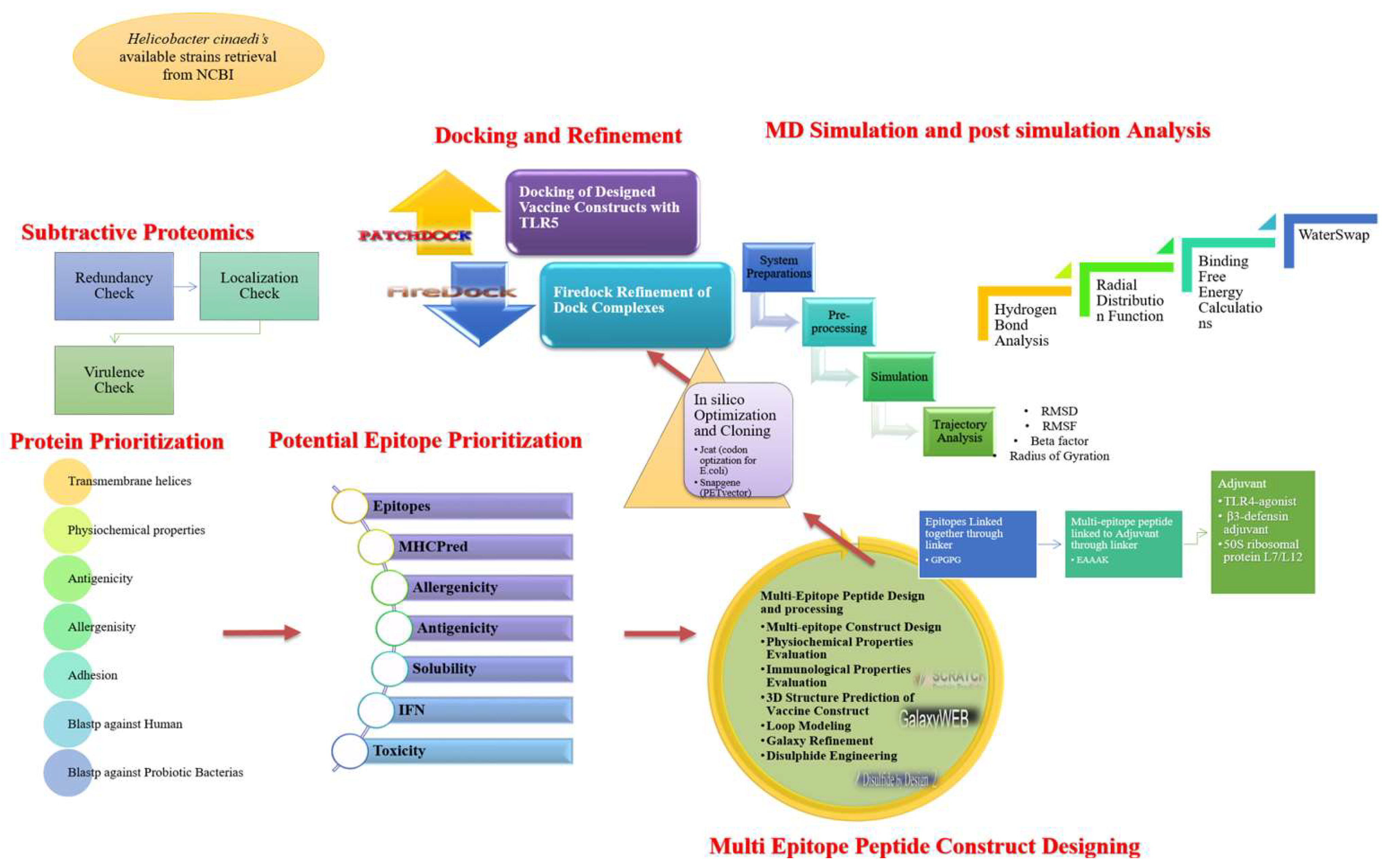
2. Methodology
2.1. Gene Analysis and Pan-Genome Exploration
2.2. Pre-Screening Phase
2.3. Prioritization of Vaccine Candidates
2.4. Multi-Epitopes’ Peptide Design
2.5. Host Immune System Simulation
2.6. Designed Vaccine Docking
2.7. Vaccine–TLR5 Dynamics Analysis
2.8. Estimation of TLR5–Vaccine Free Energies
3. Results and Discussion
3.1. Vaccine Targets’ Identification
3.2. Epitopes’ Prediction
3.3. Physicochemical Properties of MEPVC
3.4. Vaccine Structure Prediction
Disulfide Engineering
3.5. In Silico Cloning
3.6. Simulating Host Immune System
3.7. Vaccine Docking with TLR5
3.8. Molecular Dynamic Simulation
3.9. Hydrogen Bond Analysis
3.10. Binding Energy Calculations
3.11. WaterSwap Energies’ Calculation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caniça, M.; Manageiro, V.; Abriouel, H.; Moran-Gilad, J.; Franz, C.M.A.P. Antibiotic Resistance in Foodborne Bacteria. Trends Food Sci. Technol. 2019, 84, 41–44. [Google Scholar] [CrossRef]
- Antimicrobial Resistance and the Role of Vaccines | PNAS. Available online: https://www.pnas.org/content/115/51/12868.short (accessed on 21 January 2022).
- MacLean, R.C.; San Millan, A. The Evolution of Antibiotic Resistance. Science 2019, 365, 1082–1083. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 2: Management Strategies and New Agents. Pharm. Ther. 2015, 40, 344. [Google Scholar]
- The White House. National Strategy for Combating Antibiotic Resistant Bacteria; The White House-Office of the Press Secretary: Washington, DC, USA, 2014. [Google Scholar]
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.; Black, D.S.; Walsh, W.R.; Kumar, N. A New Era of Antibiotics: The Clinical Potential of Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef]
- The White House. National Action Plan for Combating Antibiotic-Resistant Bacteria; The White House-Office of the Press Secretary: Washington, DC, USA, 2015; p. 62. [Google Scholar]
- National Institutes of Health. NIAID’s Antibacterial Resistance Program: Current Status and Future Directions; NIAID: Washington, DC, USA, 2014; Volume 1, p. 13.
- Gagneux-Brunon, A.; Lucht, F.; Launay, O.; Berthelot, P.; Botelho-Nevers, E. Vaccines for Healthcare-Associated Infections: Present, Future, and Expectations. Expert Rev. Vaccines 2018, 17, 421–433. [Google Scholar] [CrossRef]
- Testerman, T.L.; Morris, J. Beyond the Stomach: An Updated View of Helicobacter Pylori Pathogenesis, Diagnosis, and Treatment. World J. Gastroenterol. WJG 2014, 20, 12781. [Google Scholar] [CrossRef]
- Solnick, J.V.; Schauer, D.B. Emergence of Diverse HelicobacterSpecies in the Pathogenesis of Gastric and Enterohepatic Diseases. Clin. Microbiol. Rev. 2001, 14, 57–59. [Google Scholar] [CrossRef]
- Kawamura, Y.; Tomida, J.; Morita, Y.; Fujii, S.; Okamoto, T.; Akaike, T. Clinical and Bacteriological Characteristics of Helicobacter Cinaedi Infection. J. Infect. Chemother. 2014, 20, 517–526. [Google Scholar] [CrossRef]
- Flahou, B.; Haesebrouck, F.; Smet, A.; Yonezawa, H.; Osaki, T.; Kamiya, S. Gastric and Enterohepatic Non-Helicobacter Pylori Helicobacters. Helicobacter 2013, 18, 66–72. [Google Scholar] [CrossRef]
- Suzuki, T.; Kutsuna, S.; Tsuboi, M.; Ota, M.; Hayakawa, K.; Ohmagari, N. Helicobacter Cinaedi Hepatic Cyst Infection with Bacteremia. Emerg. Infect. Dis. 2019, 25, 603. [Google Scholar] [CrossRef]
- Orlicek, S.L.; Welch, D.F.; Kuhls, T.L. Septicemia and Meningitis Caused by Helicobacter Cinaedi in a Neonate. J. Clin. Microbiol. 1993, 31, 569–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménard, A.; Smet, A. Review: Other Helicobacter Species. Helicobacter 2019, 24, e12645. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Hayashi, H.; Kodama, S.; Mukai, T.; Morita, Y. Bacteremia Possibly Caused by Helicobacter Cinaedi and Associated with Painful Erythema in Rheumatoid Arthritis with Malignant Lymphoma. Intern. Med. 2018, 57, 3663–3666. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.L.; Ørsted, I.; Tarpgaard, I.H.; Nielsen, H.L. Helicobacter Cinaedi Bacteraemia Secondary to Enterocolitis in an Immunocompetent Patient. Gut Pathog. 2021, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Araoka, H.; Baba, M.; Okada, C.; Kimura, M.; Sato, T.; Yatomi, Y.; Moriya, K.; Yoneyama, A. Risk Factors for Recurrent Helicobacter Cinaedi Bacteremia and the Efficacy of Selective Digestive Decontamination with Kanamycin to Prevent Recurrence. Clin. Infect. Dis. 2018, 67, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvant—An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an Ultra-Fast Pan-Genome Analysis Pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Blast, N. Basic Local Alignment Search Tool. Natl. Libr. Med. Natl. Cent. Biotechnol. Inf. 2015, 14, 1–9. [Google Scholar]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A Reference Database for Bacterial Virulence Factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved Protein Subcellular Localization Prediction with Refined Localization Subcategories and Predictive Capabilities for All Prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Yu, C.-S.; Cheng, C.-W.; Su, W.-C.; Chang, K.-C.; Huang, S.-W.; Hwang, J.-K.; Lu, C.-H. CELLO2GO: A Web Server for Protein SubCELlular LOcalization Prediction with Functional Gene Ontology Annotation. PLoS ONE 2014, 9, e99368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ProtParam, E. ExPASy-ProtParam Tool. 2017. Available online: http://life.nthu.edu.tw/~b861625/protparam (accessed on 3 January 2022).
- Sheth, H.B.; Glasier, L.M.; Ellert, N.W.; Cachia, P.; Kohn, W.; Lee, K.K.; Paranchych, W.; Hodges, R.S.; Irvin, R.T. Development of an Anti-Adhesive Vaccine for Pseudomonas Aeruginosa Targeting the C-Terminal Region of the Pilin Structural Protein. Biomed. Pept. Proteins Nucleic Acids Struct. Synth. Biol. Act. 1995, 1, 141–148. [Google Scholar]
- He, Y.; Xiang, Z.; Mobley, H.L.T. Vaxign: The First Web-Based Vaccine Design Program for Reverse Vaccinology and Applications for Vaccine Development. BioMed. Res. Int. 2010, 2010, 297505. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Humarán, L.G.; Salinas, E.; Ortiz, G.G.; Ramirez-Jirano, L.J.; Morales, J.A.; Bitzer-Quintero, O.K. From Probiotics to Psychobiotics: Live Beneficial Bacteria Which Act on the Brain-Gut Axis. Nutrients 2019, 11, 890. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Guan, P.; Doytchinova, I.A.; Zygouri, C.; Flower, D.R. MHCPred: A Server for Quantitative Prediction of Peptide–MHC Binding. Nucleic Acids Res. 2003, 31, 3621–3624. [Google Scholar] [CrossRef]
- Garg, A.; Gupta, D. VirulentPred: A SVM Based Prediction Method for Virulent Proteins in Bacterial Pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A Server for in Silico Prediction of Allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef] [PubMed]
- IFNepitope: A Server for Predicting and Designing IFN-Gamma Inducing Epitopes. Available online: http://crdd.osdd.net/raghava/ifnepitope/ (accessed on 16 August 2022).
- Nezafat, N.; Karimi, Z.; Eslami, M.; Mohkam, M.; Zandian, S.; Ghasemi, Y. Designing an Efficient Multi-Epitope Peptide Vaccine against Vibrio Cholerae via Combined Immunoinformatics and Protein Interaction Based Approaches. Comput. Biol. Chem. 2016, 62, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Meza, B.; Ascencio, F.; Sierra-Beltrán, A.P.; Torres, J.; Angulo, C. A Novel Design of a Multi-Antigenic, Multistage and Multi-Epitope Vaccine against Helicobacter Pylori: An In Silico Approach. Infect. Genet. Evol. 2017, 49, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, K.J.; Royal, J.M.; Hamorsky, K.T.; Matoba, N. Cholera Toxin B: One Subunit with Many Pharmaceutical Applications. Toxins 2015, 7, 974–996. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A Protein Structure and Structural Feature Prediction Server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast Interaction Refinement in Molecular Docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- BIOVIA Discovery Studio. Discovery Studio Visualizer; BIOVIA Discovery Studio: San Diego, CA, USA, 2017; p. 936. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cerutti, D.S.; Cheateham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; et al. AMBER16 Package; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An Accessory Software Package for Molecular Mechanical Calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Brice, A.R.; Dominy, B.N. Examining Electrostatic Influences on Base-Flipping: A Comparison of TIP3P and GB Solvent Models. Commun. Comput. Phys. 2013, 13, 223–237. [Google Scholar] [CrossRef]
- Schafmeister, C.; Ross, W.S.; Romanovski, V. The Leap Module of AMBER; University of California: San Francisco, CA, USA, 1995. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. The FF14SB Force Field. Amber 2014, 14, 29–31. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Lemak, A.S.; Balabaev, N.K. On the Berendsen Thermostat. Mol. Simul. 1994, 13, 177–187. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A Fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Sanami, S.; Zandi, M.; Pourhossein, B.; Mobini, G.R.; Safaei, M.; Abed, A.; Arvejeh, P.M.; Chermahini, F.A.; Alizadeh, M. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int. J. Biol. Macromol. 2020, 164, 871–883. [Google Scholar] [CrossRef]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Potocnakova, L.; Bhide, M.; Pulzova, L.B. An Introduction to B-Cell Epitope Mapping and in Silico Epitope Prediction. J. Immunol. Res. 2016, 2016, 6760830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, R.Y.; Ansari, A.A.; Lian, Z.X.; Gershwin, M.E. Regulatory T cells: Development, function and role in autoimmunity. Autoimmun. Rev. 2005, 4, 351–363. [Google Scholar] [CrossRef]
- Collins, M.M.; Tang, T.; Slack, R.; Sintasath, D.; Hartzman, R.J.; Ng, J.; Hurley, C.K. The Relative Frequencies of HLA-DRB1*01 Alleles in the Major US Populations. Tissue Antigens 2000, 55, 48–52. [Google Scholar] [CrossRef]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; Tahir ul Qamar, M. Pan-vaccinomics approach towards a universal vaccine candidate against WHO priority pathogens to address growing global antibiotic resistance. Comput. Biol. Med. 2021, 136, 104705. [Google Scholar] [CrossRef]
- Hooft, R.W.W.; Sander, C.; Vriend, G. Objectively Judging the Quality of a Protein Structure from a Ramachandran Plot. Bioinformatics 1997, 13, 425–430. [Google Scholar] [CrossRef]
- Creighton, T.E. Disulphide Bonds and Protein Stability. BioEssays 1988, 8, 57–63. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category of Proteins | Transmembrane Helices (TMHMM) | Physicochemical Properties | Antigenicity | Allergenicity | Adhesion | Human Blast | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino Acid | Molecular Weight | Gravy | Aliphatic Index | Instability Index | Theoretical PI | ||||||
| Outer membrane | |||||||||||
| >core/100/1/Org1_Gene1402 (TonB dependent receptor) | 1 | 726 | 81.20 | −0.42 | 76.17 | 40.6 | 8.75 | 0.60 | Non | 0.68 | Non-significant |
| Extracellular | |||||||||||
| >core/105/1/Org1_Gene1184 (Flagellar hook protein, FlgE) | 0 | 718 | 77.15 | −0.31 | 76.89 | 25.11 | 5.04 | 0.63 | Non | 0.82 | Non-significant |
| >core/1610/1/Org1_Gene663 (Hcp family type VI secretion system effector) | 0 | 171 | 18.85 | −0.46 | 74.74 | 41.28 | 5.46 | 0.79 | Non | 0.59 | Non-significant |
| Inner membrane | |||||||||||
| >core/1187/1/Org1_Gene21 (Flagellar motor protein MotB) | 1 | 250 | 27.68 | −0.28 | 86.28 | 44.78 | 4.75 | 0.63 | Non | 0.50 | Non-significant |
| Protein | B-CELL | MHC II | P. Rank | MHC I | P. Rank | MHC-Pred | Score | Allergenicity | Antigenicity | Solubility | IFN | Toxinpred |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| >core/100/1/Org1_Gene1402 | IMSELPIELQSKQI SVVEKKDLLQK | IMSELPIELQSKQIS | 12.1 | LPIELQSKQI | 0.07 | LPIELQSKQ | 34.2 | Non | Antigen | Soluble | Positive | Non-Toxin |
| GRNTLELNTLDPY | GRNTLELNTLDPY | 2.8 | RNTLELNTL | 7 | RNTLELNTL | 10.5 | Non | Antigen | Soluble | Positive | Non-Toxin | |
| SSYNTQSDTFAATARIRALEYGS VNNLFGGRAEVLGGGGG | SDTFAATARIRALEY | 9.1 | ATARIRALEY | TARIRALEY | 5.14 | Non | Antigen | Soluble | Positive | Non-Toxin | ||
| YTKDSTRYYTQGR YTRVESHRAGGLG AAPGSSYGILMSE DPISEY | RAGGLGAAPGSSYGI | 6.4 | RAGGLGAAP | 11 | RAGGLGAAP | 98 | Non | Antigen | Soluble | Positive | Non-Toxin | |
| QGRYTRVESHRAGGL | 7.9 | RYTRVESHR | 0.19 | RYTRVESHR | 9.2 | Non | Antigen | Soluble | Positive | Non-Toxin | ||
| GMKKSQLSFAESME | GMKKSQLSFAESME | 9.8 | GMKKSQLSF | 0.01 | GMKKSQLSF | 76 | Non | Antigen | Soluble | Positive | Non-Toxin | |
| TISKTKSSTEQSNNNQAI HIENSRLLDENSVIHSGA | QSNNNQAIHIENSRL | 1.2 | QAIHIENSR | 0.04 | QAIHIENSR | 12.7 | Non | Antigen | Soluble | Positive | Non-Toxin | |
| NFKNPSAGTRMQVTP SGSSTLTIANPLIKP | KNPSAGTRMQVTPSG | 3.7 | NPSAGTRMQV | 0.36 | PSAGTRMQV | 86.1 | Non | Antigen | Soluble | Positive | Non-Toxin | |
| RGYGAKTRIDPNEE RATQAYTMT | IDPNEERATQAYTMT | 20 | NEERATQAY | 0.06 | NEERATQAY | 23.1 | Non | Antigen | Soluble | Positive | Non-Toxin | |
| >core/105/1/Org1_Gene1184 | TVGFKYSRASFV | TVGFKYSRASFV | 1.3 | TVGFKYSRA | 3.3 | TVGFKYSRA | 77.6 | Non | Antigen | Soluble | Positive | Non-Toxin |
| QGWVRPPLEAAESGTMSD FDFFRVDNTGPVRNIQIDP GMVMPARATKTITLRANL NAGRHIDQMQEIAALDST ARTAADGVAPVYDSRGVLMQ | FDFFRVDNTGPVRNI | 7.6 | FDFFRVDNT | 15 | FDFFRVDNT | 24 | Non | Antigen | Soluble | Positive | Non-Toxin | |
| KTITLRANLNAGRHI | 0.79 | KTITLRANL | 0.5 | KTITLRANL | 6.7 | Non | Antigen | Soluble | Positive | Non-Toxin | ||
| AALDSTARTAADGVA | 6 | STARTAADGV | 0.69 | STARTAADG | 63.2 | Non | Antigen | Soluble | Positive | Non-Toxin | ||
| AFRYRYTKSEDADSTTGQF RTTEDLRALIQYDANM IKNPEKNYQESTASVAV | AFRYRYTKSEDADST | 6.9 | RYRYTKSEDA | 1.7 | RYRYTKSED | 12.4 | Non | Antigen | Soluble | Positive | Non-Toxin | |
| NPEKNYQESTASVAV | 9.6 | QESTASVAV | 0.13 | QESTASVAV | 21.8 | Non | Antigen | Soluble | Positive | Non-Toxin |
| Solution No. | Score | Area | Atomic Contact Energy | Transformation |
|---|---|---|---|---|
| 1 | 23,492 | 3959.3 | 113.97 | 3.06 0.83 −2.27 81.63 43.88 59.96 |
| 2 | 20,000 | 3297.6 | 123.73 | 0.83 0.47 2.98 111.75 118.06 137.80 |
| 3 | 19,574 | 3202.6 | −374.66 | −2.85 0.25 −0.50 167.15 133.05 74.02 |
| 4 | 19,538 | 2663.1 | 198.87 | −1.60 0.59 −2.94 108.59 1.32 137.65 |
| 5 | 19,298 | 4046.2 | −470.53 | 1.04 −1.25 −0.94 54.91 39.29 111.78 |
| 6 | 18,774 | 3557.9 | 100.44 | −1.03 0.12 −0.90 123.16 105.31 143.35 |
| 7 | 18,682 | 2483.8 | 429.62 | 2.85 −0.58 −2.65 118.04 91.65 57.75 |
| 8 | 18,588 | 2610.5 | 242.81 | 0.26 −0.30 −1.31 71.64 60.30 139.08 |
| 9 | 18,408 | 3281.2 | −107.39 | 2.61 0.60 −1.66 88.02 60.20 61.85 |
| 10 | 18,264 | 3714.8 | 353.54 | −1.37 0.59 2.51 65.97 19.97 137.43 |
| 11 | 18,174 | 3498.7 | −227.03 | 2.12 0.23 −0.99 75.52 54.46 52.60 |
| 12 | 18,080 | 2482.3 | 234.36 | 0.12 −0.59 −1.43 76.73 53.89 133.59 |
| 13 | 17,852 | 2598.1 | 430.16 | −1.31 0.64 1.86 62.13 59.69 124.78 |
| 14 | 17,564 | 2876.2 | 166.54 | −2.56 0.22 −2.35 97.84 68.44 93.37 |
| 15 | 17,558 | 3028.1 | 95.72 | −3.13 0.94 −2.51 75.37 44.11 61.76 |
| 16 | 17,494 | 2829.5 | 243.03 | 2.91 0.39 0.29 111.91 80.87 72.90 |
| 17 | 17,466 | 2496 | 376.63 | −0.02 0.60 0.18 81.40 43.87 182.40 |
| 18 | 17,434 | 2112.7 | 412.27 | 1.90 −0.36 0.69 114.91 56.10 60.34 |
| 19 | 17,408 | 2299.3 | 456.21 | −0.88 −0.19 2.51 88.98 92.68 116.39 |
| 20 | 17,276 | 2243.5 | 162.58 | 2.76 −0.86 −0.46 49.05 99.23 33.79 |
| Rank | Solution Number | Global Energy | Attractive van der Waals Energy | Repulsive van der Waals Energy | Atomic Contact Energy | Hydrogen Bonding Energy |
|---|---|---|---|---|---|---|
| 1 | 3 | −13.7 | −13.19 | 5.03 | −7.61 | −0.83 |
| 2 | 10 | 8.40 | −14.75 | 13.32 | 8.68 | −1.62 |
| 3 | 2 | 12.38 | −9.09 | 3.37 | 7.04 | −2.23 |
| 4 | 9 | 13.54 | −11.29 | 18.74 | 6.08 | −1.61 |
| 5 | 8 | 30.08 | −19.11 | 58.67 | 9.00 | −2.15 |
| 6 | 4 | 64.05 | −18.81 | 101.92 | 14.73 | −3.36 |
| 7 | 1 | 165.6 | −20.49 | 203.14 | 8.92 | −1.40 |
| 8 | 5 | 200.3 | −26.97 | 236.17 | 3.87 | −1.96 |
| 9 | 7 | 652.5 | −33.22 | 873.36 | 15.34 | −7.18 |
| 10 | 6 | 1300.1 | −39.65 | 1620.54 | 20.31 | −7.70 |
| Energy Parameter | TLR5–Vaccine Complex |
|---|---|
| MMGBSA | |
| Van der Waals (ΔEvdw) | −391.03 |
| Electrostatic (ΔEele) | −156.97 |
| Polar (ΔGsolv/GB) | 88.99 |
| Non-polar (ΔGnpol) | −20.87 |
| Gas phase | −548 |
| Solvation | 68.12 |
| Net (tot/GB) | −479.88 |
| MMPBSA | |
| Van der Waals (ΔEvdw) | −391.03 |
| Electrostatic (ΔEele) | −156.97 |
| Polar (ΔGsolv/PB) | 87.63 |
| Non-polar (ΔGnpol) | −23.78 |
| Gas phase | −548 |
| Solvation | 63.85 |
| Net (Δtot/PB) | −484.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, S.; Alsowayeh, N.; Abbasi, H.W.; Albutti, A.; Tahir ul Qamar, M.; Ahmad, S.; Raza, R.Z.; Sadia, K.; Abbasi, S.W. Pan-Genome-Assisted Computational Design of a Multi-Epitopes-Based Vaccine Candidate against Helicobacter cinaedi. Int. J. Environ. Res. Public Health 2022, 19, 11579. https://doi.org/10.3390/ijerph191811579
Ismail S, Alsowayeh N, Abbasi HW, Albutti A, Tahir ul Qamar M, Ahmad S, Raza RZ, Sadia K, Abbasi SW. Pan-Genome-Assisted Computational Design of a Multi-Epitopes-Based Vaccine Candidate against Helicobacter cinaedi. International Journal of Environmental Research and Public Health. 2022; 19(18):11579. https://doi.org/10.3390/ijerph191811579
Chicago/Turabian StyleIsmail, Saba, Noorah Alsowayeh, Hyder Wajid Abbasi, Aqel Albutti, Muhammad Tahir ul Qamar, Sajjad Ahmad, Rabail Zehra Raza, Khulah Sadia, and Sumra Wajid Abbasi. 2022. "Pan-Genome-Assisted Computational Design of a Multi-Epitopes-Based Vaccine Candidate against Helicobacter cinaedi" International Journal of Environmental Research and Public Health 19, no. 18: 11579. https://doi.org/10.3390/ijerph191811579
APA StyleIsmail, S., Alsowayeh, N., Abbasi, H. W., Albutti, A., Tahir ul Qamar, M., Ahmad, S., Raza, R. Z., Sadia, K., & Abbasi, S. W. (2022). Pan-Genome-Assisted Computational Design of a Multi-Epitopes-Based Vaccine Candidate against Helicobacter cinaedi. International Journal of Environmental Research and Public Health, 19(18), 11579. https://doi.org/10.3390/ijerph191811579