
Decoding Diabetes Biomarkers and Related Molecular Mechanisms by Using Machine Learning, Text Mining, and Gene Expression Analysis

 ,
,  ,
,  , , , and
, , , and
Abstract
:1. Introduction
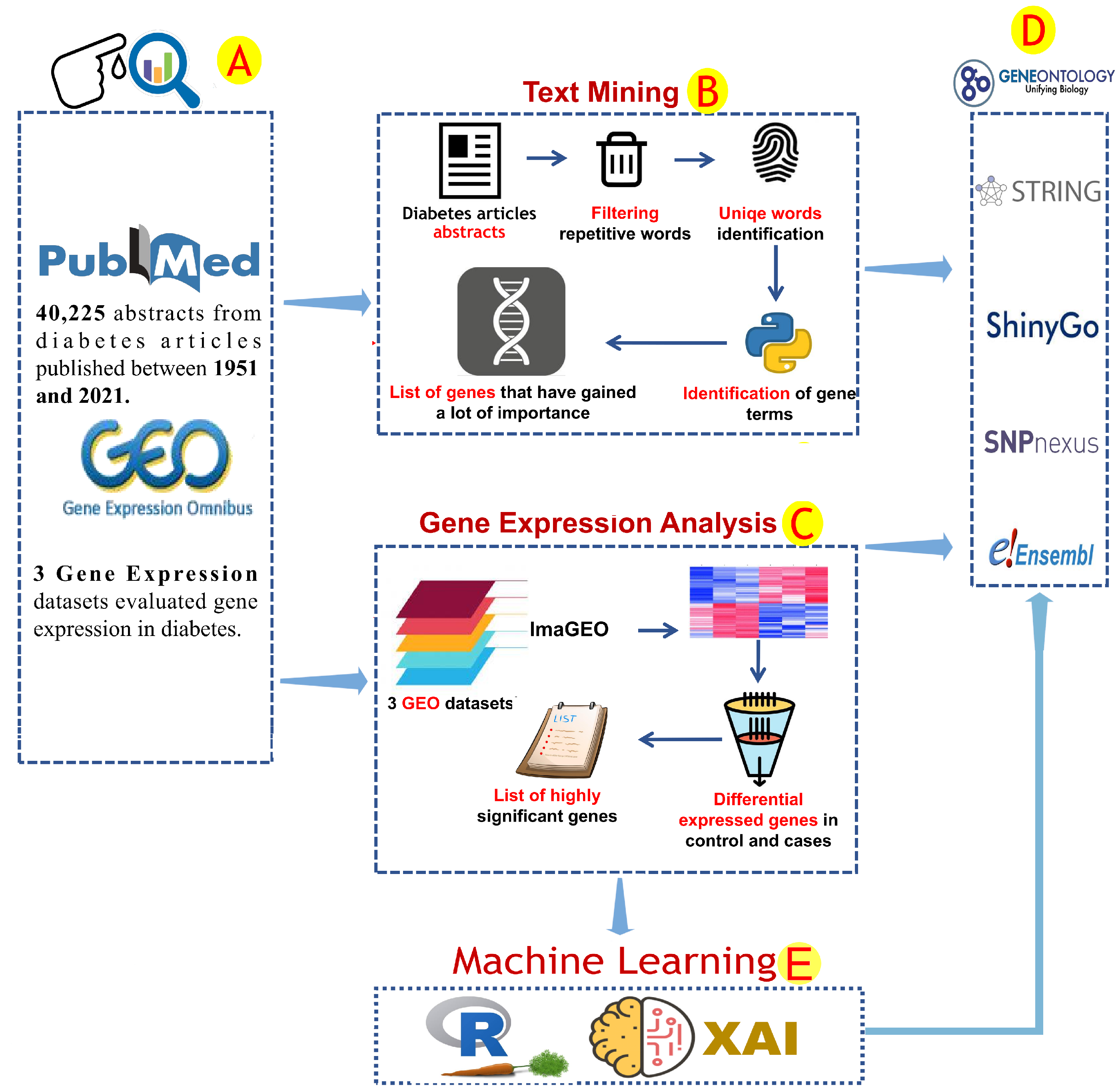
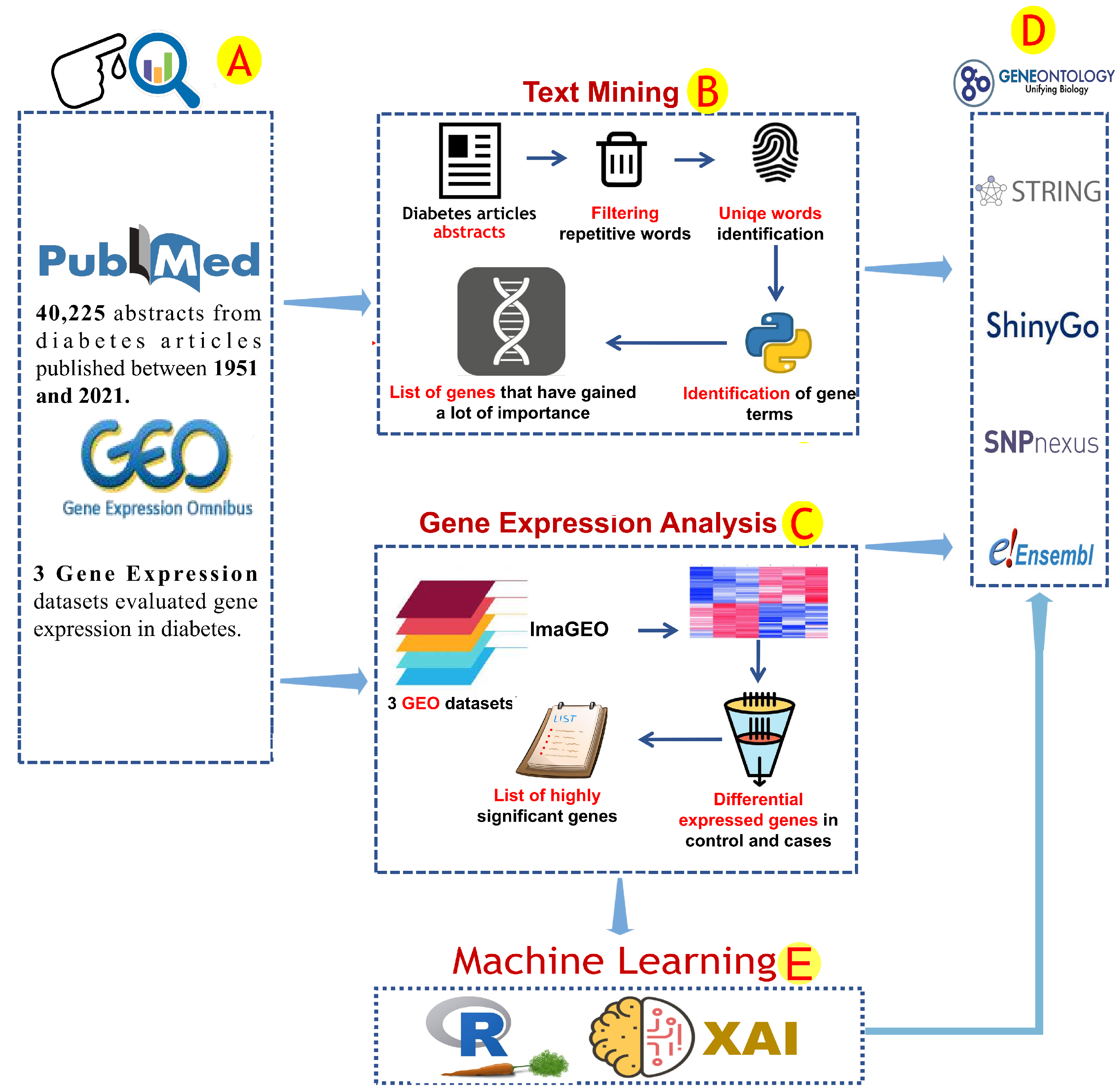
2. Materials and Methods
2.1. Text Mining Analysis
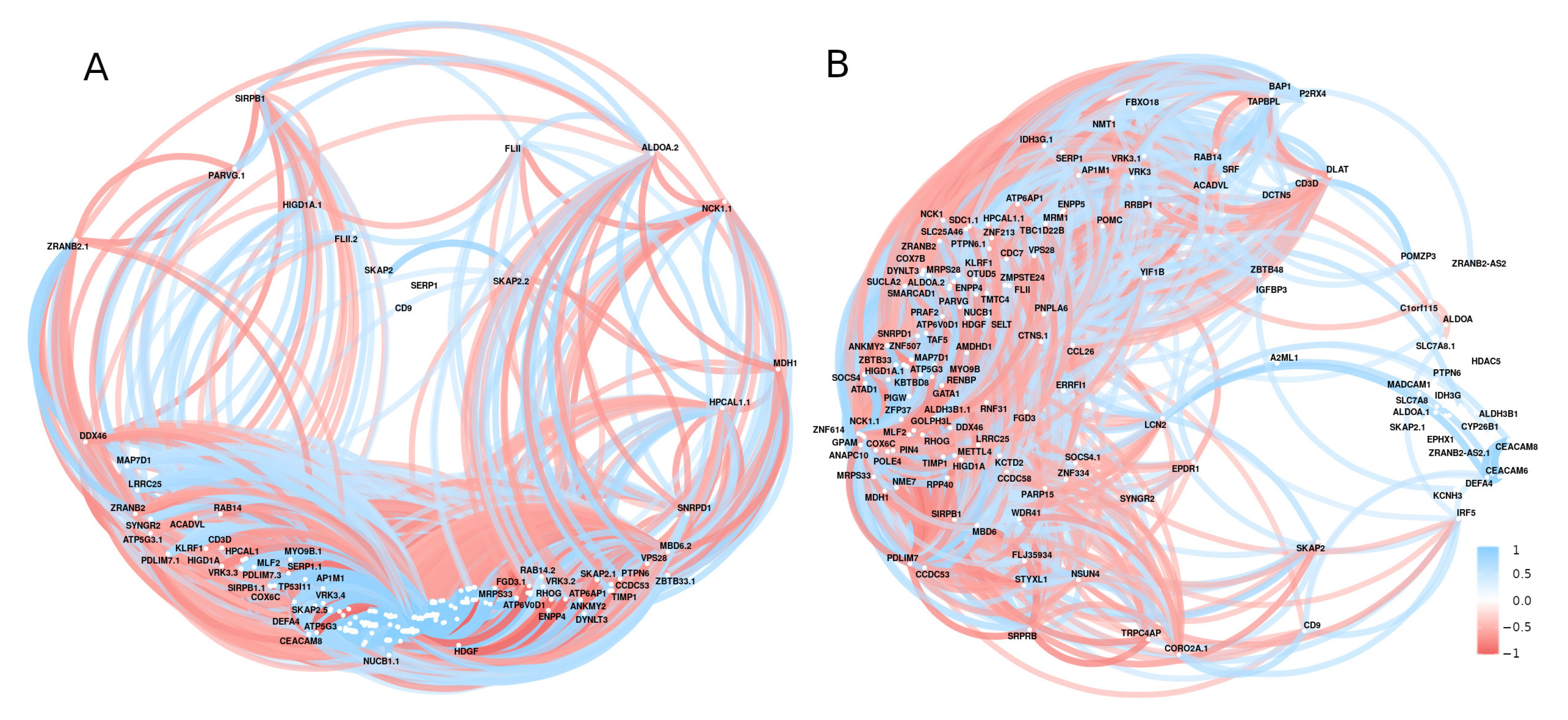
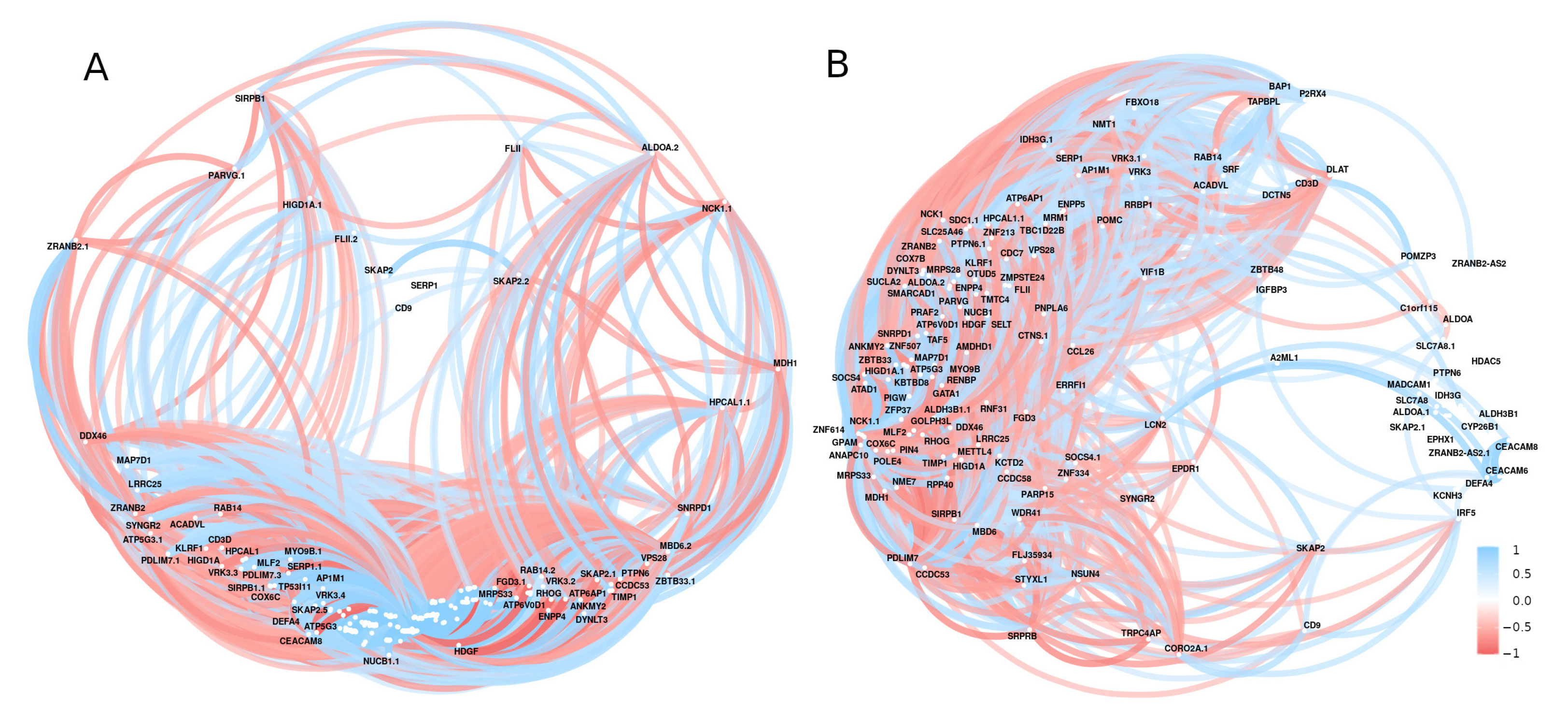
2.2. Gene Expression Analysis and Correlation Analysis
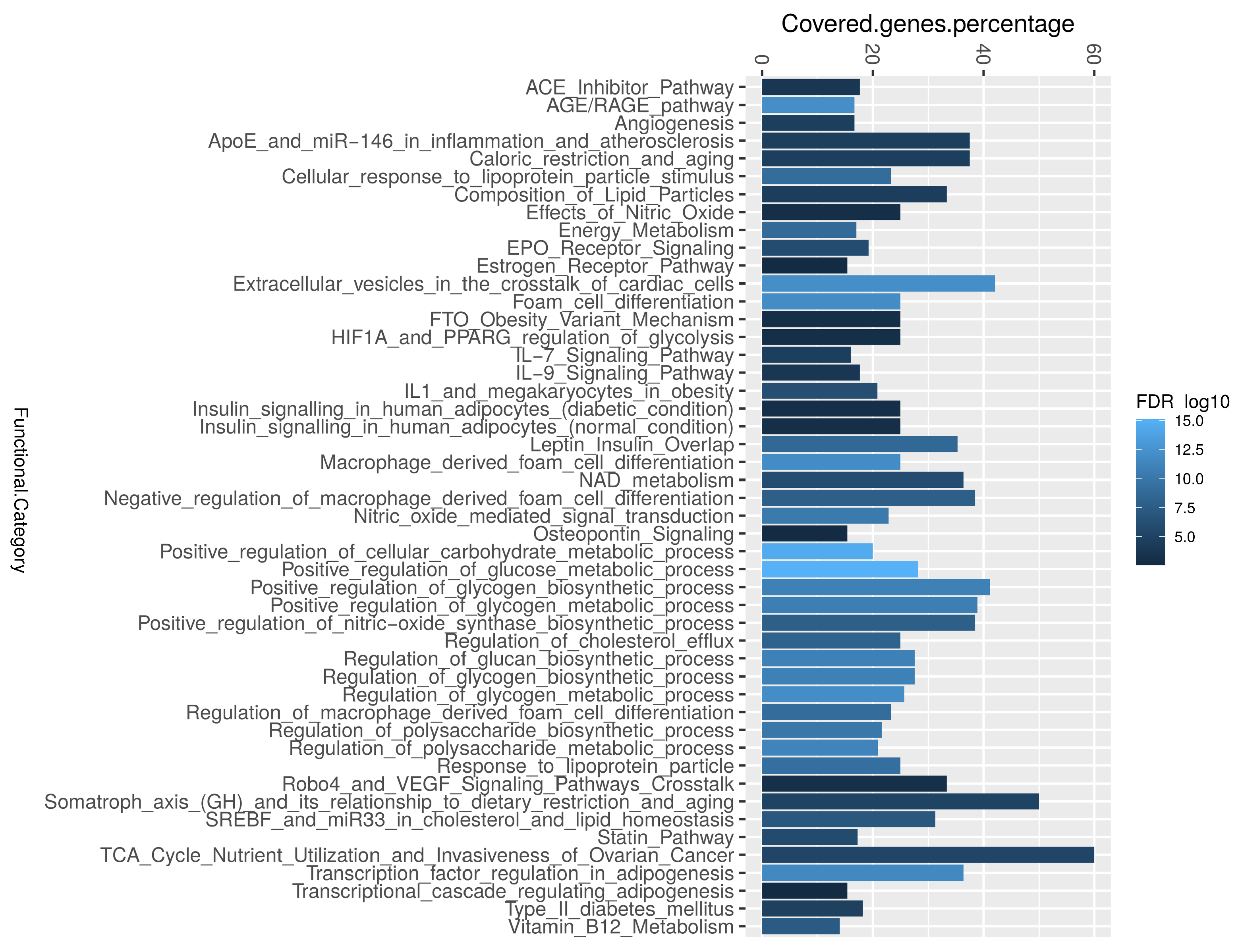
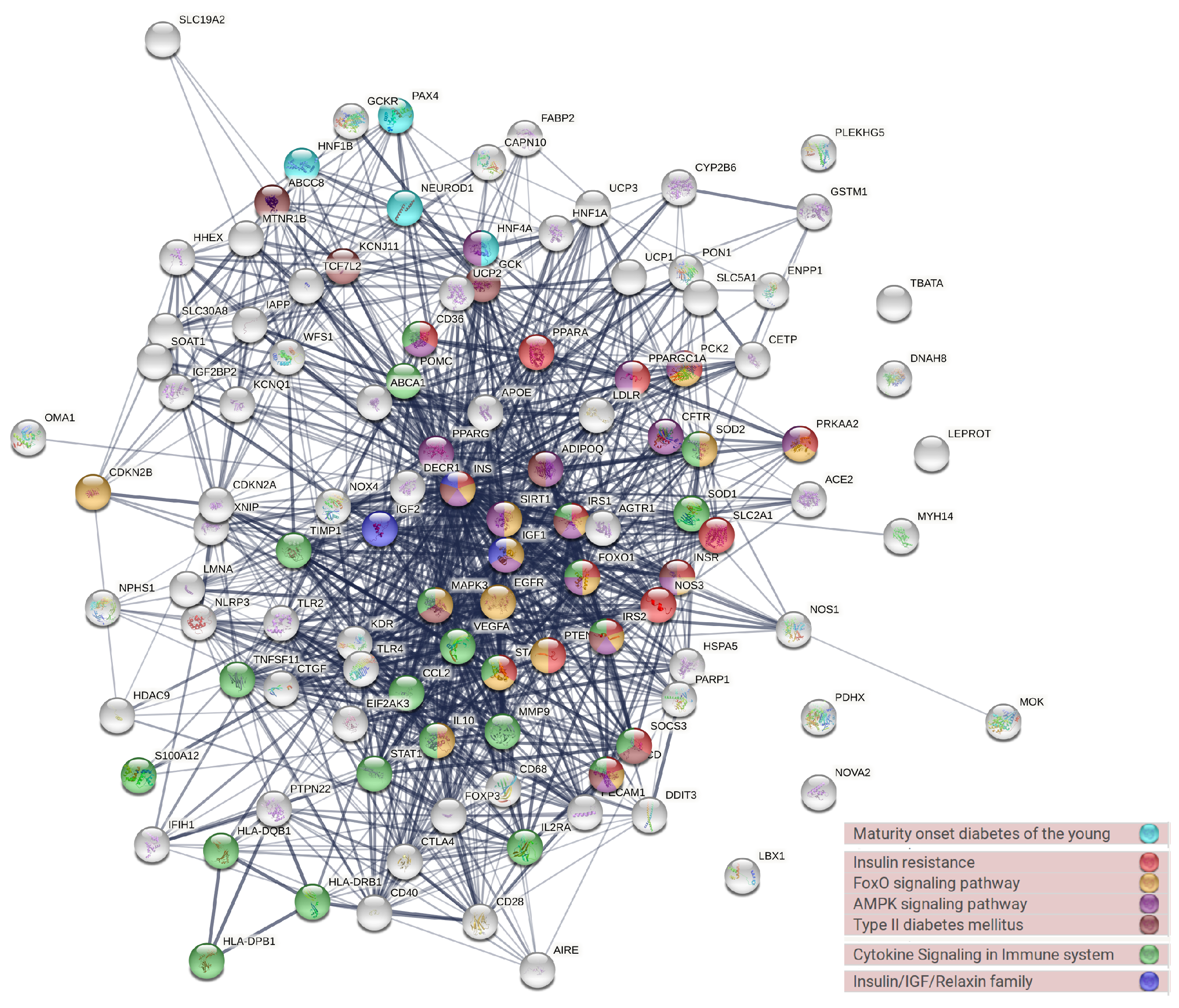
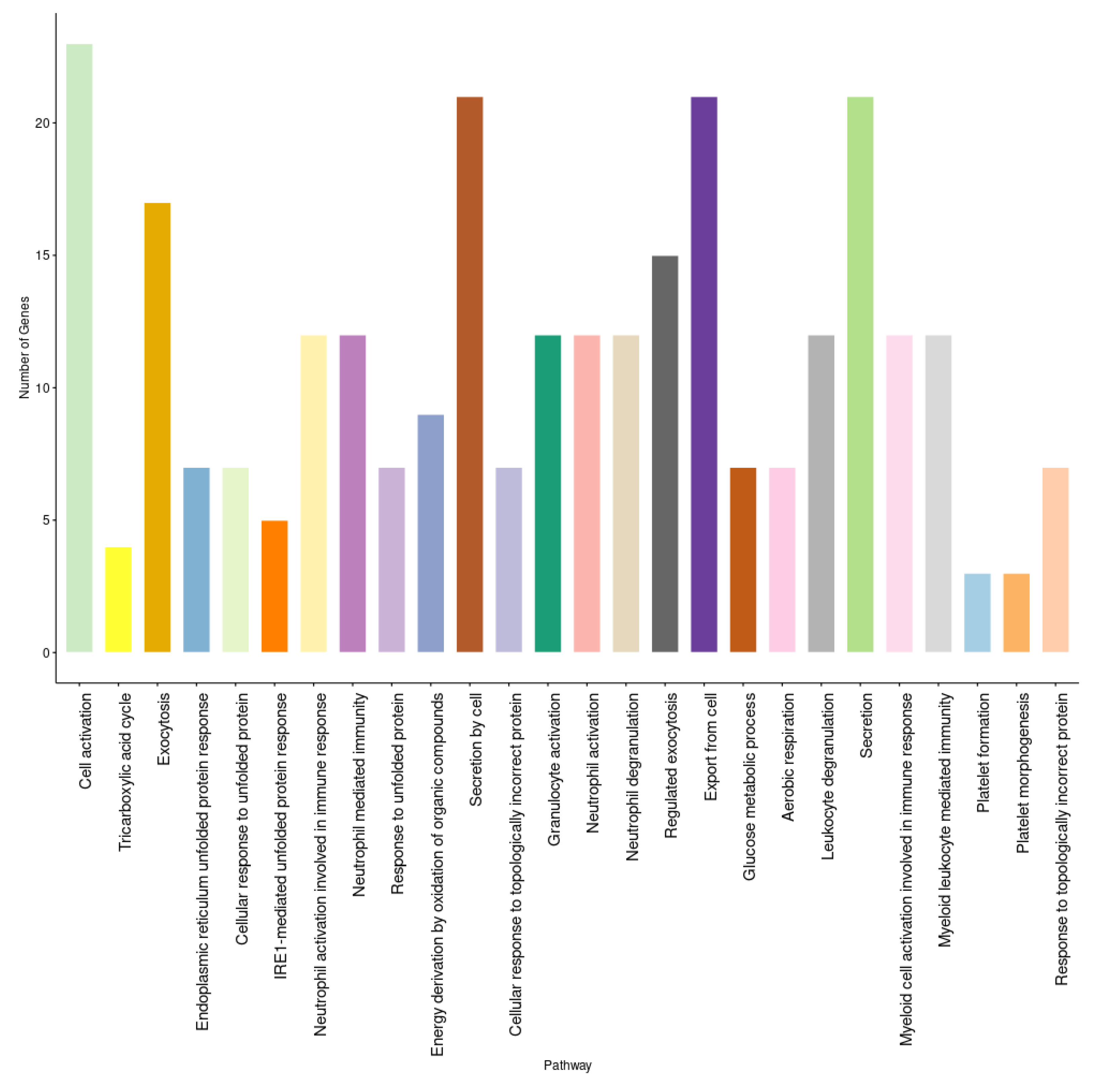
2.3. Enrichment Analysis and Protein–Protein Interactions
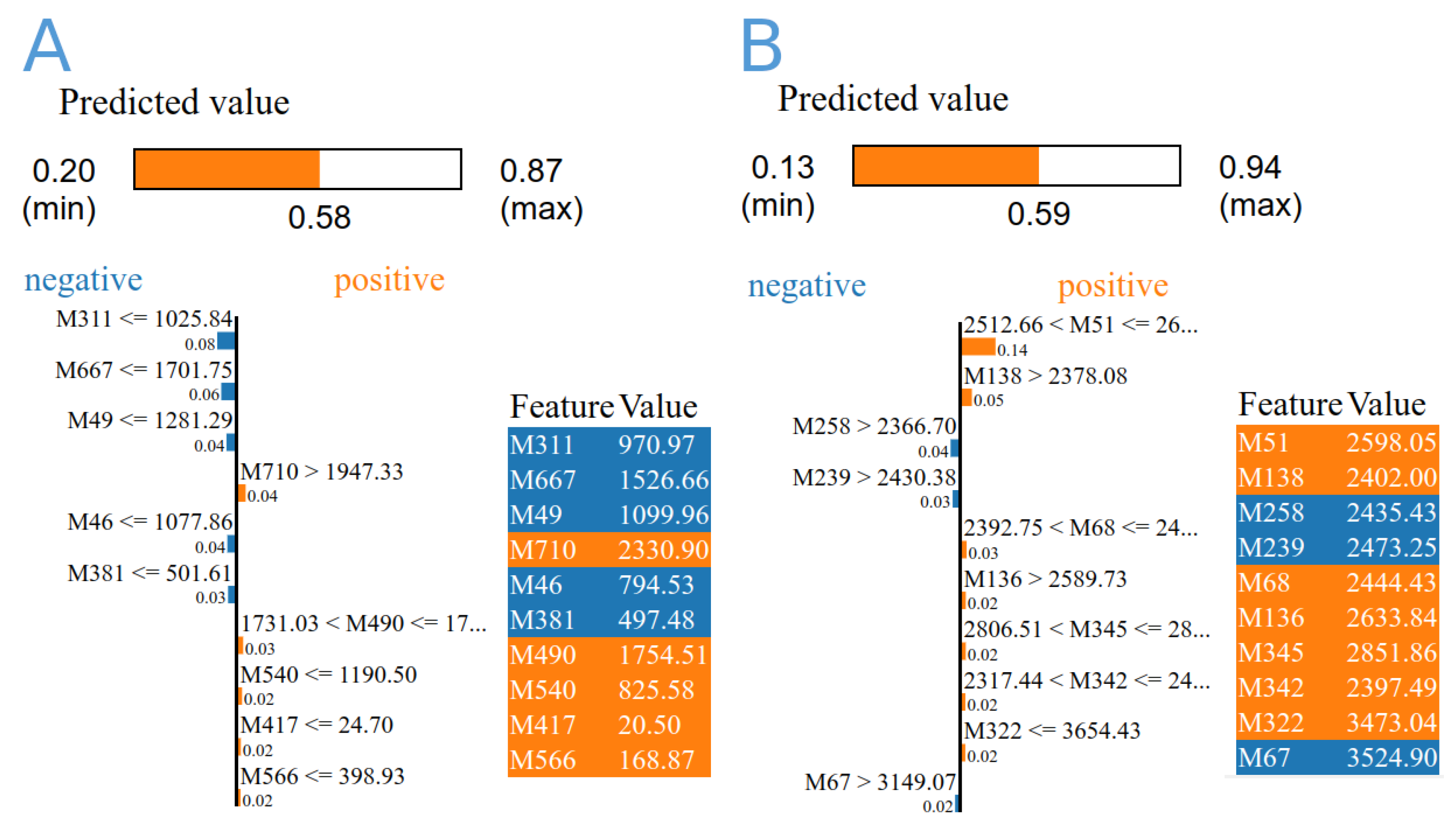
2.4. Machine Learning Analysis and Correlation Analyses
3. Results
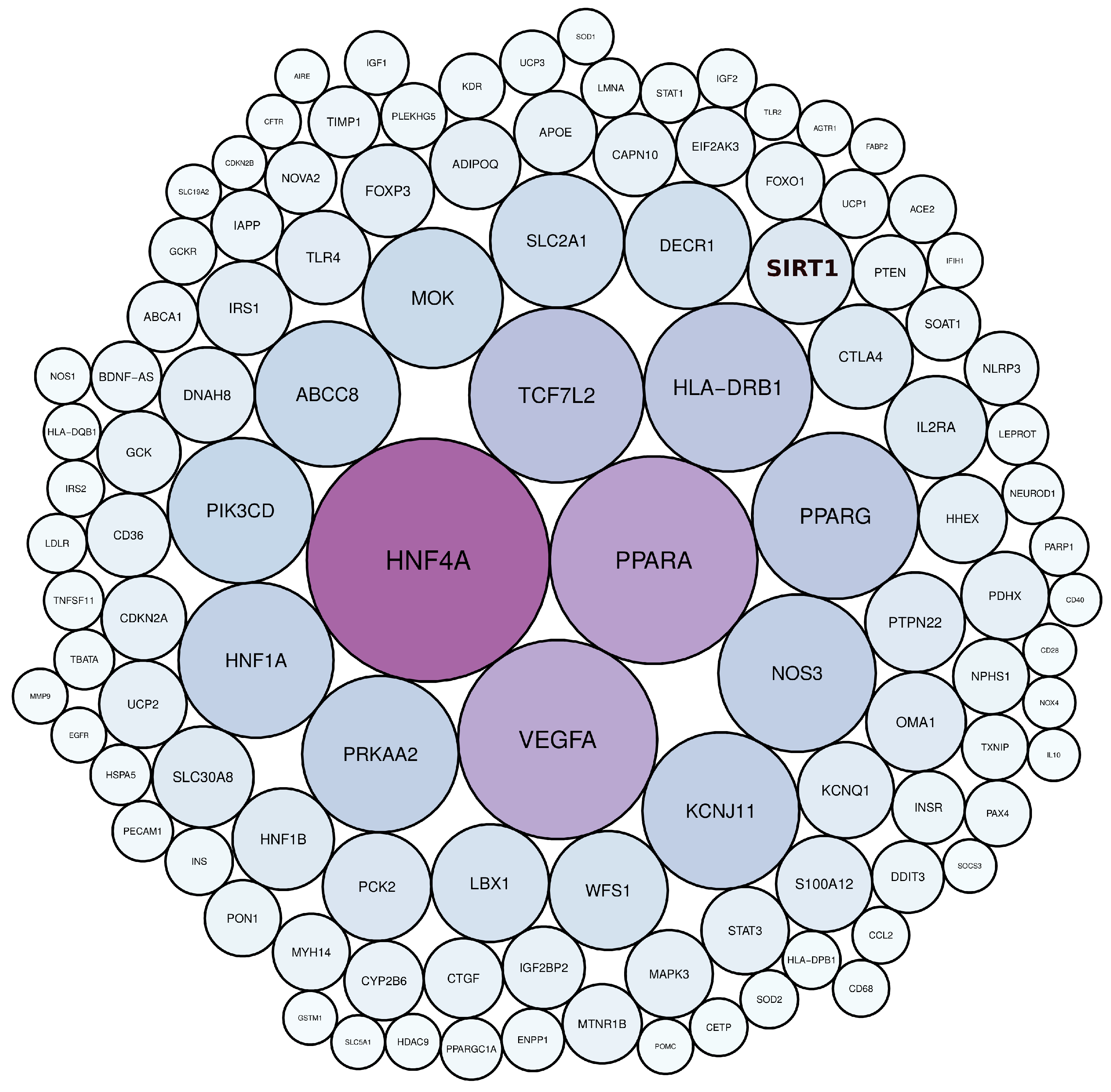
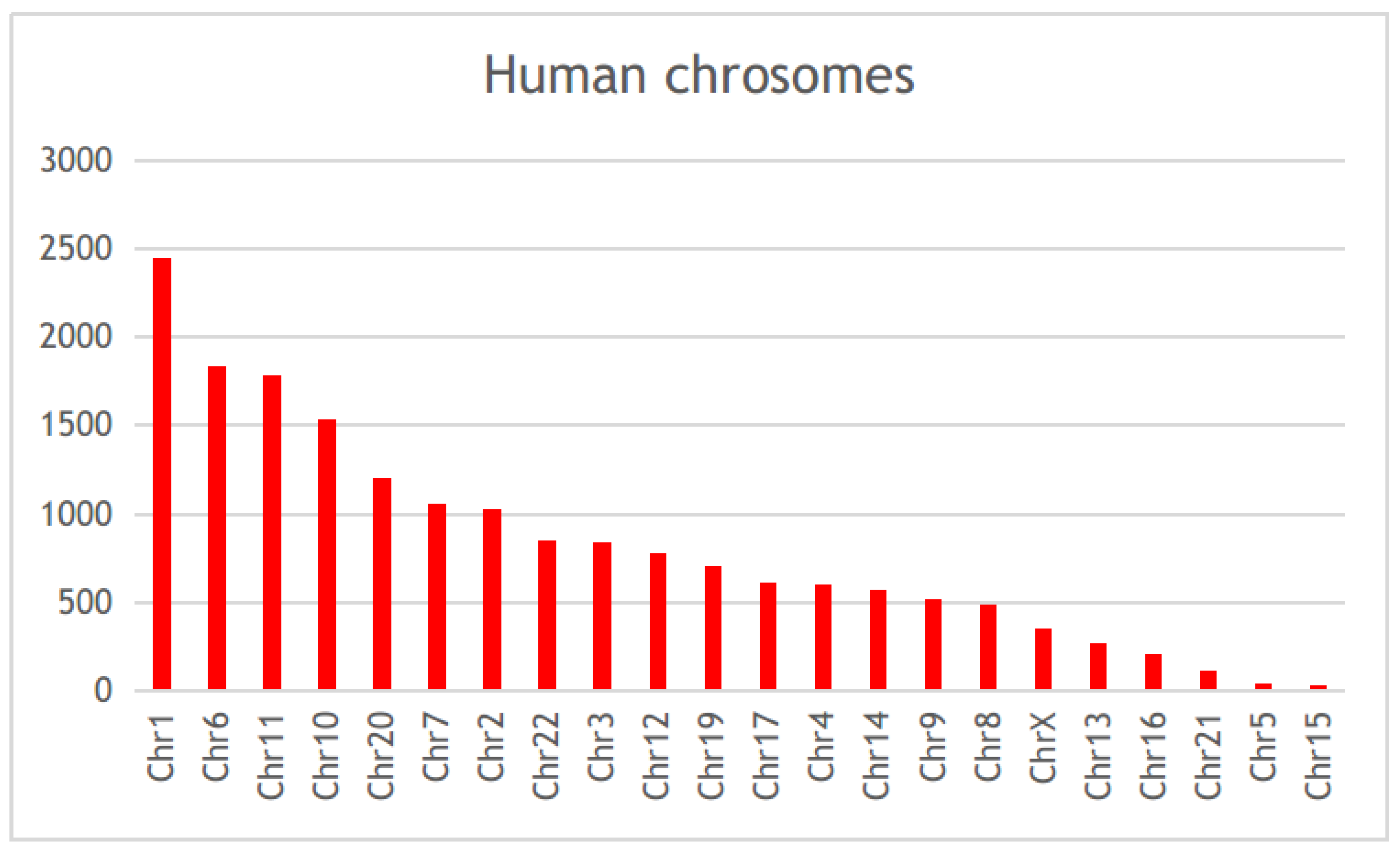
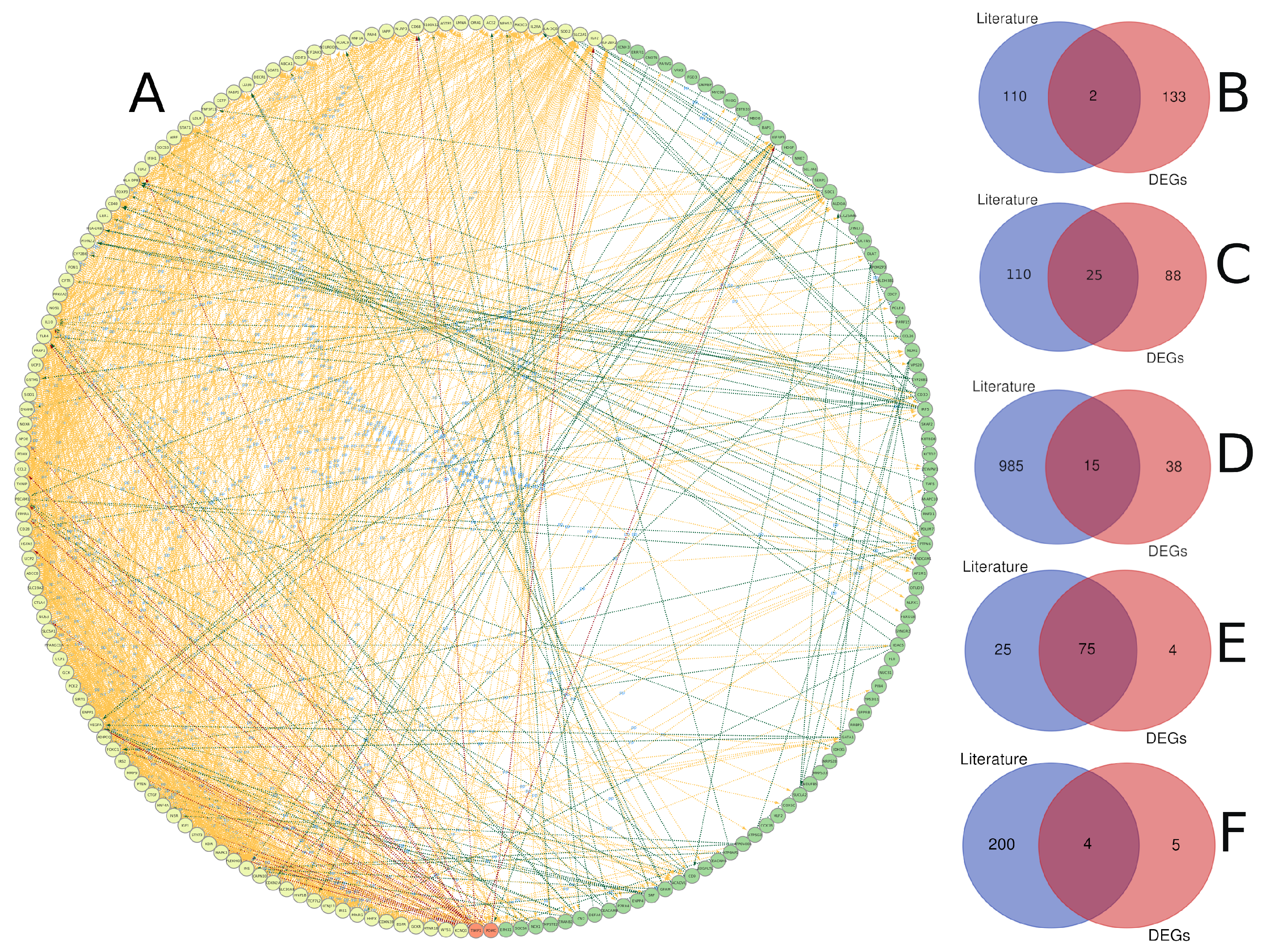
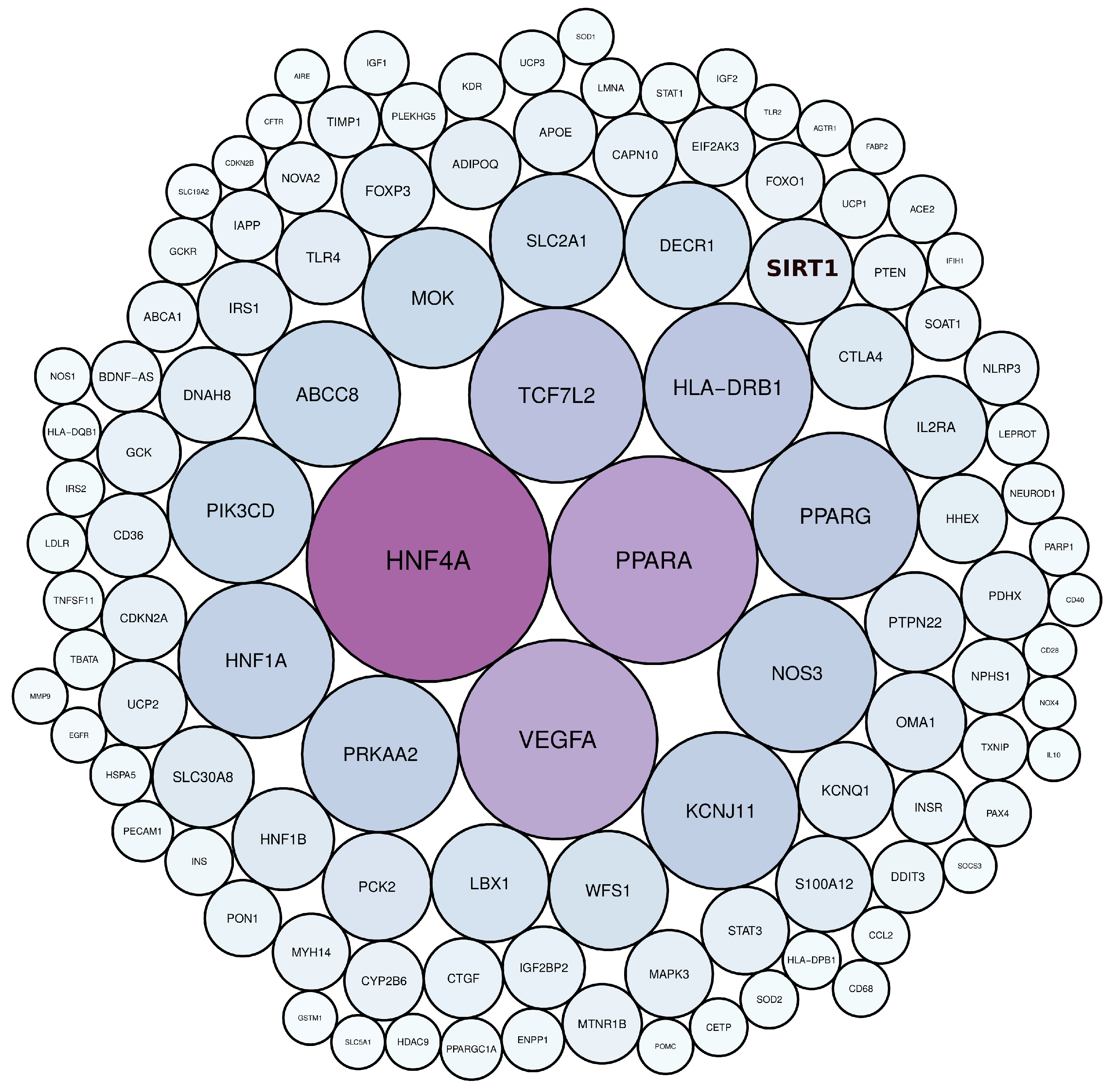
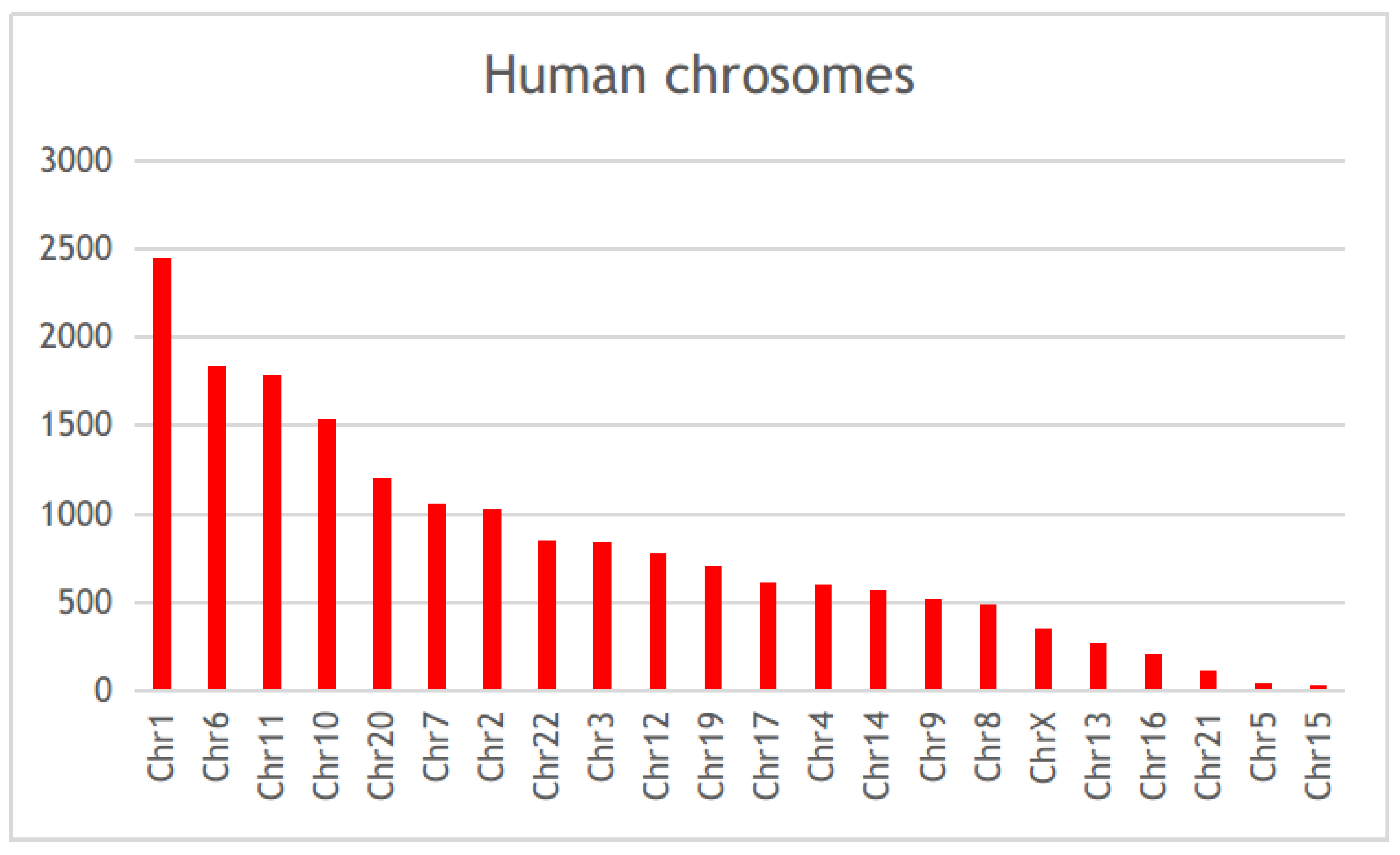
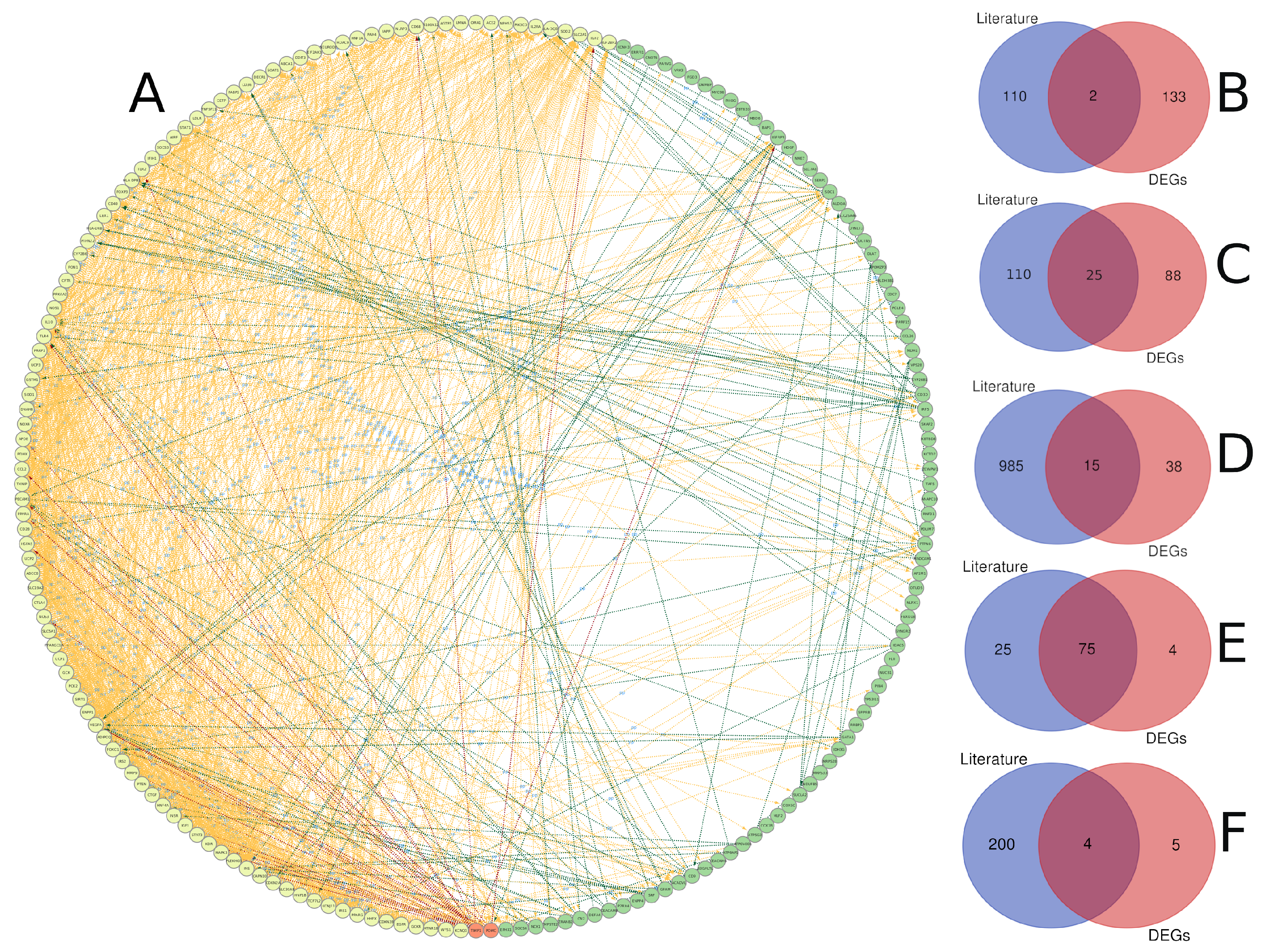
3.1. Diabetes-Related Genes Occurring Frequently in the Literature
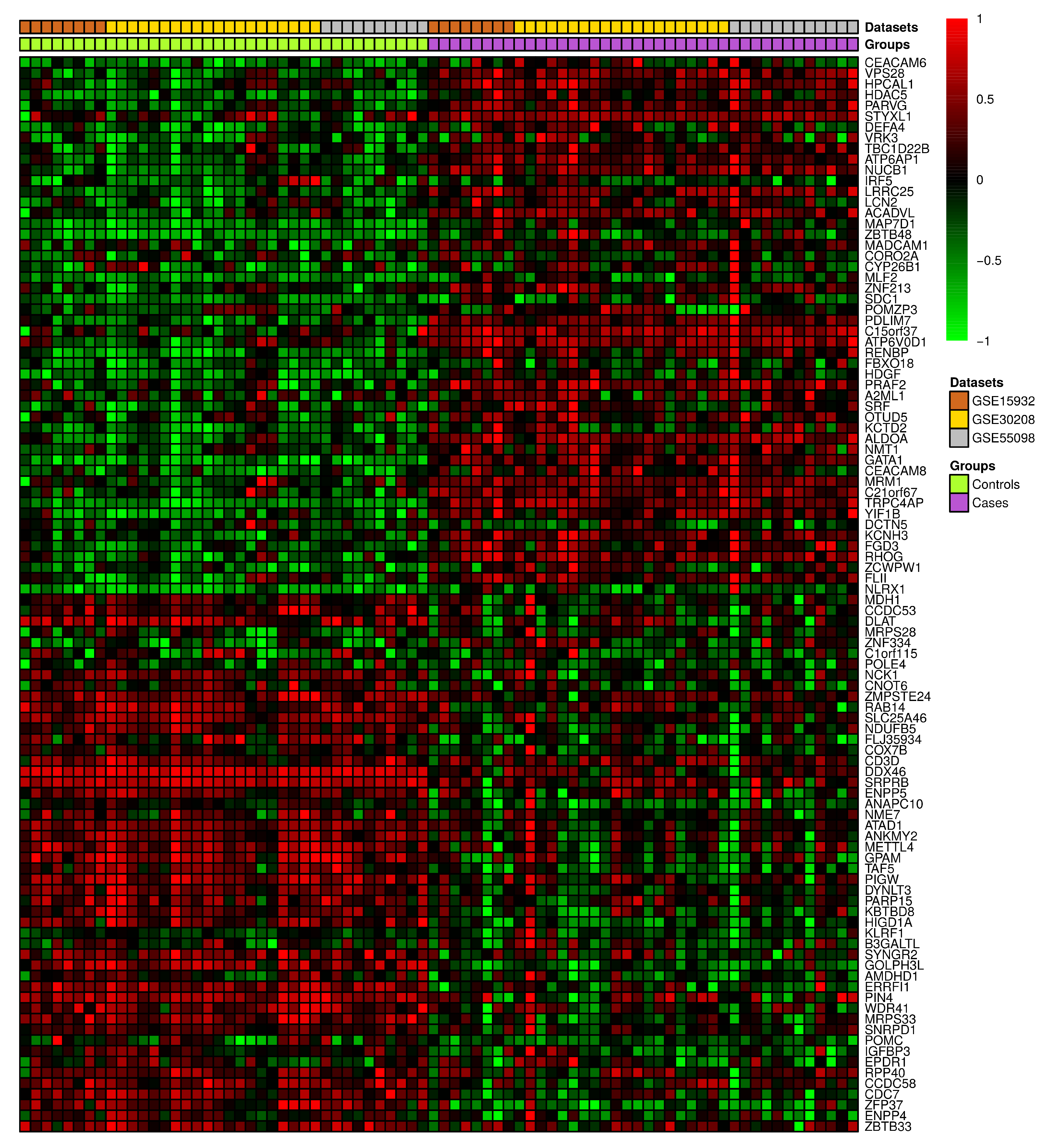
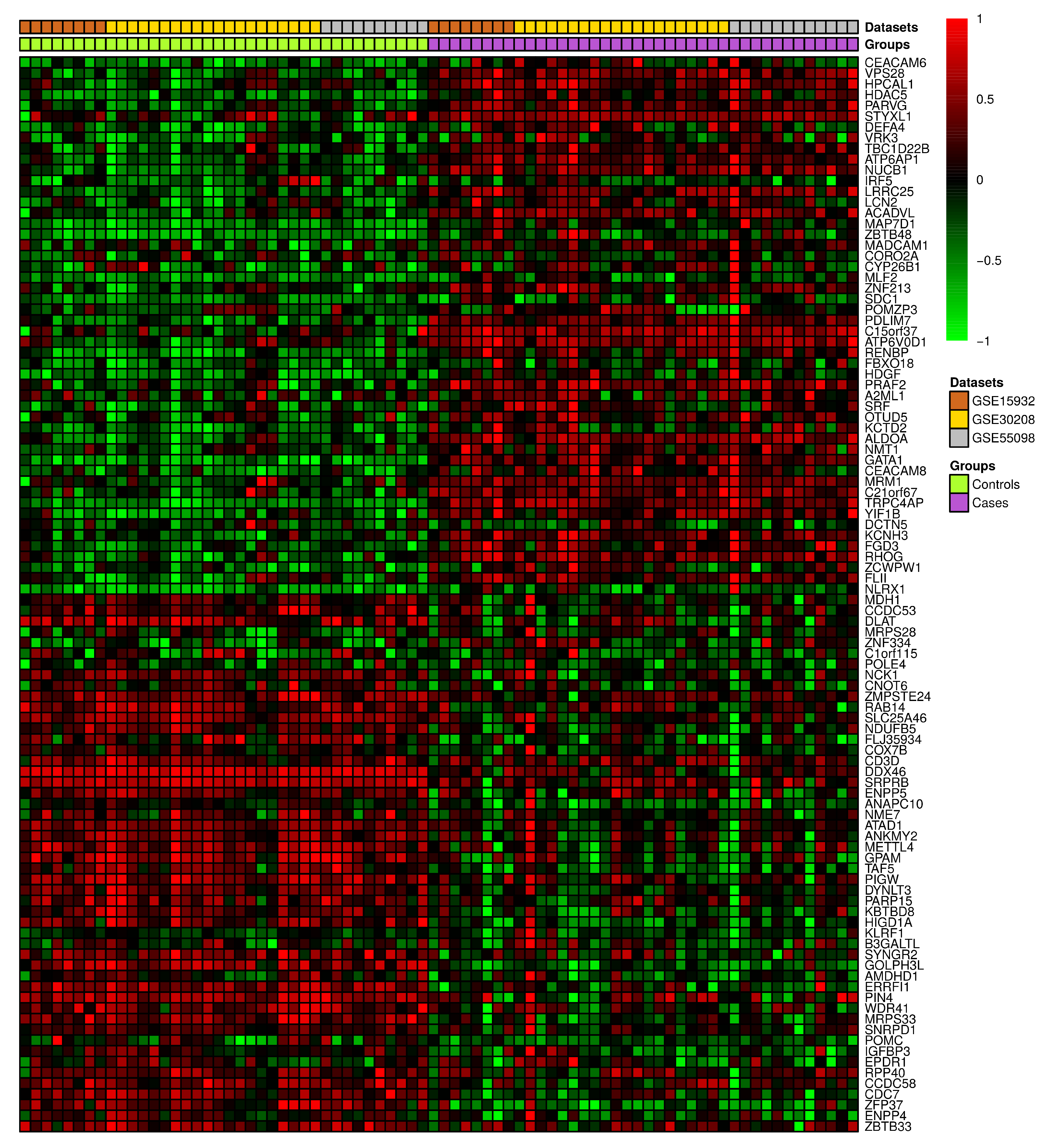
3.2. Differential Gene Expression and Correlation Analyses
3.3. Text Mining versus Gene Expression
3.4. Machine Learning Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DEG | Differential expressed genes |
| ER | Endoplasmic reticulum |
| GEO | Gene Expression Omnibus |
| IDF | International Diabetes Federation |
| PPI | Protein-protein interaction |
| TCA | Tricarboxylic acid |
| TCR | T-Cell antigen Receptor |
References
- Cefalu, W.T.; Berg, E.G.; Saraco, M.; Petersen, M.P.; Uelmen, S.; Robinson, S. Classification and diagnosis of diabetes: Standards of medical care in diabetes-2019. Diabetes Care 2019, 42, S13–S28. [Google Scholar]
- Zimmet, P.; Alberti, K.G.; Shaw, J. Global and societal implications of the diabetes epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef]
- Williams, R.; Colagiuri, S.; Almutairi, R.; Montoya, P.A.; Basit, A.; Beran, D.; Besançon, S.; Bommer, C.; Borgnakke, W.; Boyko, E.; et al. IDF Diabetes Atlas; International Diabetes Atlas: Brussels, Belgium, 2019. [Google Scholar]
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IDF Diabetes Atlas. International diabetes federation. In IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015; pp. 1–163. [Google Scholar]
- Alsamman, A.M.; Zayed, H. The transcriptomic profiling of SARS-CoV-2 compared to SARS, MERS, EBOV, and H1N1. PLoS ONE 2020, 15, e0243270. [Google Scholar] [CrossRef] [PubMed]
- Udhaya Kumar, S.; Thirumal Kumar, D.; Bithia, R.; Sankar, S.; Magesh, R.; Sidenna, M.; George Priya Doss, C.; Zayed, H. Analysis of differentially expressed genes and molecular pathways in familial hypercholesterolemia involved in atherosclerosis: A systematic and bioinformatics approach. Front. Genet. 2020, 11, 734. [Google Scholar] [CrossRef] [PubMed]
- Lipatova, A.V.; Soboleva, A.V.; Gorshkov, V.A.; Bubis, J.A.; Solovyeva, E.M.; Krasnov, G.S.; Kochetkov, D.V.; Vorobyev, P.O.; Ilina, I.Y.; Moshkovskii, S.A.; et al. Multi-Omics Analysis of Glioblastoma Cells’ Sensitivity to Oncolytic Viruses. Cancers 2021, 13, 5268. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yu, Z.; Cheng, X.; Luo, Y.; Wen, C. A bibliometric analysis and visualization of medical data mining research. Medicine 2020, 99, e20338. [Google Scholar] [CrossRef]
- Tanwar, H.; Kumar, D.T.; Doss, C.; Zayed, H. Bioinformatics classification of mutations in patients with Mucopolysaccharidosis IIIA. Metab. Brain Dis. 2019, 34, 1577–1594. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Lachmann, A.; Ma’ayan, A. Mining data and metadata from the gene expression omnibus. Biophys. Rev. 2019, 11, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Dang, M.; Li, H.; Jin, X.; Yang, W. Identification of genes related to mental disorders by text mining. Medicine 2019, 98, e17504. [Google Scholar] [CrossRef]
- Lee, W.Y.; Bachtiar, M.; Choo, C.C.; Lee, C.G. Comprehensive review of H epatitis BV irus-associated hepatocellular carcinoma research through text mining and big data analytics. Biol. Rev. 2019, 94, 353–367. [Google Scholar] [CrossRef] [PubMed]
- El Naqa, I.; Murphy, M.J. What is machine learning? In Machine Learning in Radiation Oncology; Springer: Cham, Switzerland, 2015; pp. 3–11. [Google Scholar]
- Sidey-Gibbons, J.A.; Sidey-Gibbons, C.J. Machine learning in medicine: A practical introduction. BMC Med. Res. Methodol. 2019, 19, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniruzzaman, M.; Rahman, M.J.; Ahammed, B.; Abedin, M.M.; Suri, H.S.; Biswas, M.; El-Baz, A.; Bangeas, P.; Tsoulfas, G.; Suri, J.S. Statistical characterization and classification of colon microarray gene expression data using multiple machine learning paradigms. Comput. Methods Programs Biomed. 2019, 176, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Linardatos, P.; Papastefanopoulos, V.; Kotsiantis, S. Explainable ai: A review of machine learning interpretability methods. Entropy 2020, 23, 18. [Google Scholar] [CrossRef]
- Schedlbauer, J.; Raptis, G.; Ludwig, B. Medical informatics labor market analysis using web crawling, web scraping, and text mining. Int. J. Med. Inform. 2021, 150, 104453. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.; Ledoux, P.; Evangelista, C.; Kim, I.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Res 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Toro-Domínguez, D.; Martorell-Marugán, J.; López-Domínguez, R.; García-Moreno, A.; González-Rumayor, V.; Alarcón-Riquelme, M.E.; Carmona-Sáez, P. ImaGEO: Integrative gene expression meta-analysis from GEO database. Bioinformatics 2019, 35, 880–882. [Google Scholar] [CrossRef]
- Benesty, J.; Chen, J.; Huang, Y.; Cohen, I. Pearson correlation coefficient. In Noise Reduction in Speech Processing; Springer: Berlin/Heidelberg, Germany, 2009; pp. 1–4. [Google Scholar]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Bailey, T.L. DREME: Motif discovery in transcription factor ChIP-seq data. Bioinformatics 2011, 27, 1653–1659. [Google Scholar] [CrossRef] [Green Version]
- Figueira, L.; Li, W.; McWilliam, H.; Lopez, R.; Xenarios, I.; Bougueleret, L.; Bridge, A.; Poux, S.; Redaschi, N.; Aimo, L.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2; Springer: New York, NY, USA, 2009; Volume 10, pp. 978–1000. [Google Scholar]
- Alsamman, A.M.; Habib, P.T. GeneSyno: Simple tool to extract gene sequence from the human genome despite synonymous gene terms. Highlights Biosci. 2019, 2. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Kido, Y.; Biggs, W.H.; Wright, C.V.; White, M.F.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic β cell growth. J. Clin. Investig. 2002, 110, 1839–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.K.; Vatamaniuk, M.Z.; Lee, C.S.; Flaschen, R.C.; Fulmer, J.T.; Matschinsky, F.M.; Duncan, S.A.; Kaestner, K.H. The MODY1 gene HNF-4α regulates selected genes involved in insulin secretion. J. Clin. Investig. 2005, 115, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Azizi, S.M.; Sarhangi, N.; Afshari, M.; Abbasi, D.; Meybodi, H.R.A.; Hasanzad, M. Association analysis of the HNF4A common genetic variants with type 2 diabetes mellitus risk. Int. J. Mol. Cell. Med. 2019, 8, 56. [Google Scholar]
- Peixoto-Barbosa, R.; Reis, A.F.; Giuffrida, F. Update on clinical screening of maturity-onset diabetes of the young (MODY). Diabetol. Metab. Syndr. 2020, 12, 50. [Google Scholar] [CrossRef]
- Temtem, M.; Serrao, M.; Mendonca, M.; Santos, M.; Sousa, A.; Mendonca, F.; Sousa, A.; Henriques, E.; Freitas, S.; Rodrigues, M.; et al. Is HNF4A gene, a risk factor or protection against coronary artery disease? Eur. Heart J. 2021, 42, ehab724.3196. [Google Scholar] [CrossRef]
- Arvind, K.; Pradeepa, R.; Deepa, R.; Mohan, V. Diabetes and coronary artery disease. IJMR 2002, 116, 121–132. [Google Scholar]
- Beale, E.G.; Harvey, B.J.; Forest, C. PCK1 and PCK2 as candidate diabetes and obesity genes. Cell Biochem. Biophys. 2007, 48, 89–95. [Google Scholar] [CrossRef]
- Yang, J.; Kalhan, S.C.; Hanson, R.W. What is the metabolic role of phosphoenolpyruvate carboxykinase? J. Biol. Chem. 2009, 284, 27025–27029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Yan, H.; Wang, H.; Zhang, Y.; Cao, H.; Wan, Y.; Kong, L.; Chen, S.; Xu, H.; Pan, B.; et al. AQR is a novel type 2 diabetes-associated gene that regulates signaling pathways critical for glucose metabolism. J. Genet. Genom. 2018, 45, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Konopelska, S.; Kienitz, T.; Quinkler, M. Downregulation of hepatic glucose 6-phosphatase-alpha in patients with nonalcoholic fatty liver. In Endocrine Abstracts; Bioscientifica: Bristol, UK, 2009; Volume 19. [Google Scholar]
- Haeusler, R.A.; Camastra, S.; Astiarraga, B.; Nannipieri, M.; Anselmino, M.; Ferrannini, E. Decreased expression of hepatic glucokinase in type 2 diabetes. Mol. Metab. 2015, 4, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Karbasforooshan, H.; Karimi, G. The role of SIRT1 in diabetic cardiomyopathy. Biomed. Pharmacother. 2017, 90, 386–392. [Google Scholar] [CrossRef]
- Iskender, H.; Dokumacioglu, E.; Sen, T.M.; Ince, I.; Kanbay, Y.; Saral, S. The effect of hesperidin and quercetin on oxidative stress, NF-κB and SIRT1 levels in a STZ-induced experimental diabetes model. Biomed. Pharmacother. 2017, 90, 500–508. [Google Scholar] [CrossRef]
- de Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Man, C.D.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: Potential biochemical mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Dong, H.H. FoxO integration of insulin signaling with glucose and lipid metabolism. J. Endocrinol. 2017, 233, R67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, J.A.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Downes, K.; Plagnol, V.; Bailey, R.; Nejentsev, S.; Field, S.F.; Payne, F.; et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007, 39, 857–864. [Google Scholar] [CrossRef]
- Pandey, R.; Zhou, M.; Islam, S.; Chen, B.; Barker, N.K.; Langlais, P.; Srivastava, A.; Luo, M.; Cooke, L.S.; Weterings, E.; et al. Carcinoembryonic antigen cell adhesion molecule 6 (CEACAM6) in Pancreatic Ductal Adenocarcinoma (PDA): An integrative analysis of a novel therapeutic target. Sci. Rep. 2019, 9, 18347. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Li, J.; Lv, S.; Yu, J.; Jiang, J.; Yao, J.; Xiao, Y.; Xu, B.; He, H.; Guo, F.; et al. HDAC5 integrates ER stress and fasting signals to regulate hepatic fatty acid oxidation. J. Lipid Res. 2018, 59, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Jia, K.; Wang, H.; Gao, F.; Zhao, S.; Li, F.; Hao, J. METTL14-regulated PI3K/Akt signaling pathway via PTEN affects HDAC5-mediated epithelial–mesenchymal transition of renal tubular cells in diabetic kidney disease. Cell Death Dis. 2021, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Olaniyi, K.S.; Amusa, O.A.; Ajadi, I.O.; Alabi, B.Y.; Agunbiade, T.B.; Ajadi, M.B. Repression of HDAC5 by acetate restores hypothalamic-pituitary-ovarian function in type 2 diabetes mellitus. Reprod. Toxicol. 2021, 106, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Mattei, A.M.; Smailys, J.D.; Hepworth, E.M.W.; Hinton, S.D. The roles of pseudophosphatases in disease. Int. J. Mol. Sci. 2021, 22, 6924. [Google Scholar] [CrossRef] [PubMed]
- Scheeff, E.D.; Eswaran, J.; Bunkoczi, G.; Knapp, S.; Manning, G. Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure 2009, 17, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Buvall, L.; Rashmi, P.; Lopez-Rivera, E.; Andreeva, S.; Weins, A.; Wallentin, H.; Greka, A.; Mundel, P. Proteasomal degradation of Nck1 but not Nck2 regulates RhoA activation and actin dynamics. Nat. Commun. 2013, 4, 2863. [Google Scholar] [CrossRef] [Green Version]
- Yamani, L.; Li, B.; Larose, L. Nck1 deficiency improves pancreatic β cell survival to diabetes-relevant stresses by modulating PERK activation and signaling. Cell. Signal. 2015, 27, 2555–2567. [Google Scholar] [CrossRef]
- Lo, A.S.Y.; Liew, C.T.; Ngai, S.M.; Tsui, S.K.W.; Fung, K.P.; Lee, C.Y.; Waye, M.M.Y. Developmental regulation and cellular distribution of human cytosolic malate dehydrogenase (MDH1). J. Cell. Biochem. 2005, 94, 763–773. [Google Scholar] [CrossRef]
- Jörns, A.; Ishikawa, D.; Teraoku, H.; Yoshimoto, T.; Wedekind, D.; Lenzen, S. Remission of autoimmune diabetes by anti-TCR combination therapies with anti-IL-17A or/and anti-IL-6 in the IDDM rat model of type 1 diabetes. BMC Med. 2020, 18, 33. [Google Scholar] [CrossRef]
- Pei, Q.; Li, J.; Zhou, P.; Zhang, J.; Huang, P.; Fan, J.; Zou, Z.; Li, X.; Wang, B. A Potential Participant in Type 2 Diabetes Bone Fragility: TIMP-1 at Sites of Osteocyte Lacunar-Canalicular System. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 4903. [Google Scholar] [CrossRef]
- Goit, R.K.; Taylor, A.W.; Lo, A.C.Y. The central melanocortin system as a treatment target for obesity and diabetes: A brief overview. Eur. J. Pharmacol. 2022, 924, 174956. [Google Scholar] [CrossRef]
- Bakalov, V.K.; Cheng, C.; Zhou, J.; Bondy, C.A. X-chromosome gene dosage and the risk of diabetes in Turner syndrome. J. Clin. Endocrinol. Metab. 2009, 94, 3289–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, E.A.; Gillespie, K.M. Diabetes and gender. Diabetologia 2001, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komosinska-Vassev, K.; Olczyk, P.; Winsz-Szczotka, K.; Kuznik-Trocha, K.; Klimek, K.; Olczyk, K. Age-and gender-dependent changes in connective tissue remodeling: Physiological differences in circulating MMP-3, MMP-10, TIMP-1 and TIMP-2 level. Gerontology 2011, 57, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Li, Y.Y.; Zheng, S.G.; Tsun, A.; Li, B. FOXP3+ Treg cells and gender bias in autoimmune diseases. Front. Immunol. 2015, 6, 493. [Google Scholar] [CrossRef]
- Connor, S.C.; Hansen, M.K.; Corner, A.; Smith, R.F.; Ryan, T.E. Integration of metabolomics and transcriptomics data to aid biomarker discovery in type 2 diabetes. Mol. BioSystems 2010, 6, 909–921. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J. Am. Soc. Nephrol. 2006, 17, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Farina, F.; Picascia, S.; Pisapia, L.; Barba, P.; Vitale, S.; Franzese, A.; Mozzillo, E.; Gianfrani, C.; Del Pozzo, G.G. HLA-DQA1 and HLA-DQB1 alleles, conferring susceptibility to celiac disease and type 1 diabetes, are more expressed than non-predisposing alleles and are coordinately regulated. Cells 2019, 8, 751. [Google Scholar] [CrossRef] [Green Version]
- Simmons, K.M.; Mitchell, A.M.; Alkanani, A.A.; McDaniel, K.A.; Baschal, E.E.; Armstrong, T.; Pyle, L.; Yu, L.; Michels, A.W. Failed genetic protection: Type 1 diabetes in the presence of HLA-DQB1* 06: 02. Diabetes 2020, 69, 1763–1769. [Google Scholar] [CrossRef]
- Mäkinen, M.; Löyttyniemi, E.; Koskinen, M.; Vähä-Mäkilä, M.; Siljander, H.; Nurmio, M.; Mykkänen, J.; Virtanen, S.M.; Simell, O.; Hyöty, H.; et al. Serum 25-hydroxyvitamin D concentrations at birth in children screened for HLA-DQB1 conferred risk for type 1 diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 2277–2285. [Google Scholar] [CrossRef]
- Gerasimou, P.; Nicolaidou, V.; Skordis, N.; Picolos, M.; Monos, D.; Costeas, P.A. Combined effect of glutamine at position 70 of HLA-DRB1 and alanine at position 57 of HLA-DQB1 in type 1 diabetes: An epitope analysis. PLoS ONE 2018, 13, e0193684. [Google Scholar] [CrossRef] [Green Version]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [PubMed] [Green Version]
- Abrams, A.J.; Fontanesi, F.; Tan, N.B.; Buglo, E.; Campeanu, I.J.; Rebelo, A.P.; Kornberg, A.J.; Phelan, D.G.; Stark, Z.; Zuchner, S. Insights into the genotype-phenotype correlation and molecular function of SLC25A46. Hum. Mutat. 2018, 39, 1995–2007. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Peng, Y.; Hufnagel, R.B.; Hu, Y.C.; Zhao, C.; Queme, L.F.; Khuchua, Z.; Driver, A.M.; Dong, F.; Lu, Q.R.; et al. Loss of SLC25A46 causes neurodegeneration by affecting mitochondrial dynamics and energy production in mice. Hum. Mol. Genet. 2017, 26, 3776–3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Yanobu-Takanashi, R.; Shimizu, Y.; Takahashi, Y.; Hiura, K.; Watanabe, M.; Sasaki, H.; Okamura, T.; Sasaki, N. Genetic locus responsible for diabetic phenotype in the insulin hyposecretion (ihs) mouse. PLoS ONE 2020, 15, e0234132. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, Z.; Guo, Z.; Dai, G.; Li, Y.; Chen, Y.; Xie, R.; Wang, X.; Cui, M.; Lu, G.; et al. Preliminary Study of Genome-Wide Association Identified Novel Susceptibility Genes for Hemorheological Indexes in a Chinese Population. Transfus. Med. Hemotherapy 2022, 1–11. [Google Scholar] [CrossRef]
- Sindhu, S.; Kochumon, S.; Thomas, R.; Bennakhi, A.; Al-Mulla, F.; Ahmad, R. Enhanced adipose expression of interferon regulatory factor (IRF)-5 associates with the signatures of metabolic inflammation in diabetic obese patients. Cells 2020, 9, 730. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, H.A.; Pastar, I.; Jozic, I.; Stojadinovic, O.; Stone, R.C.; Ojeh, N.; Gil, J.; Davis, S.C.; Kirsner, R.S.; Tomic-Canic, M. Staphylococcus aureus triggers induction of miR-15B-5P to diminish DNA repair and deregulate inflammatory response in diabetic foot ulcers. J. Investig. Dermatol. 2018, 138, 1187–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, C.H.; Choi, D.S. Essential roles for the non-canonical IκB kinases in linking inflammation to cancer, obesity, and diabetes. Cells 2019, 8, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-β links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
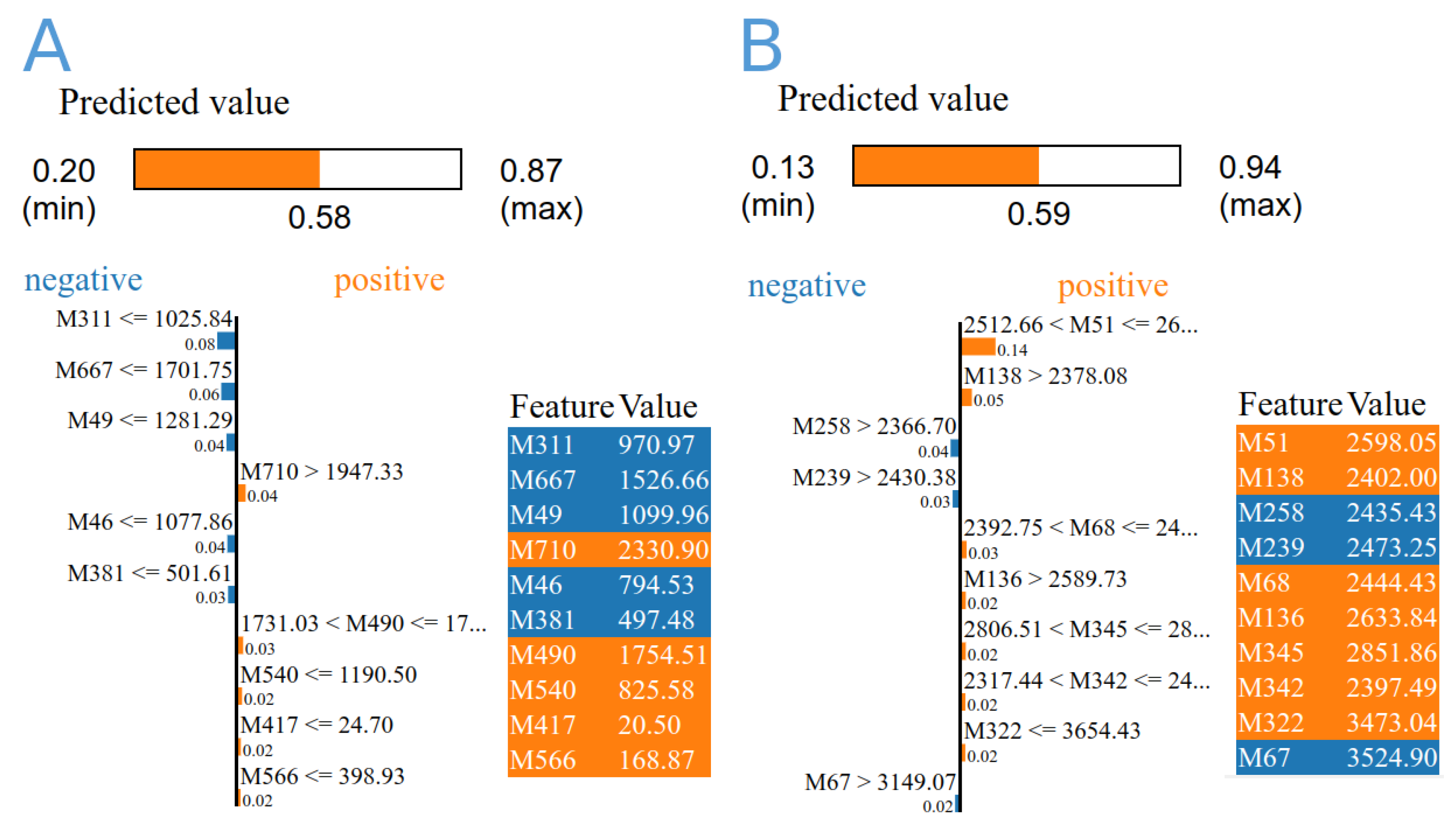{kind=link}
| Term Name | FDR | Share | Intersection | Term Name | FDR | Share | Intersection |
|---|---|---|---|---|---|---|---|
| response to nitrogen compound | 33.3 | 47.27% | 4.73% | positive regulation of macromolecule metabolic process | 19.3 | 58.18% | 1.85% |
| response to organonitrogen compound | 32.6 | 45.45% | 4.95% | positive regulation of multicellular organismal process | 19.3 | 40.00% | 3.07% |
| regulation of multicellular organismal process | 30.9 | 62.73% | 2.59% | apoptotic process | 19.2 | 44.55% | 2.63% |
| cellular response to chemical stimulus | 30.6 | 65.45% | 2.38% | localization | 19.1 | 76.36% | 1.31% |
| response to endogenous stimulus | 28 | 49.09% | 3.47% | signaling | 19.1 | 76.36% | 1.30% |
| chemical homeostasis | 27.6 | 41.82% | 4.50% | cell death | 19 | 46.36% | 2.46% |
| regulation of biological quality | 27.2 | 68.18% | 1.99% | positive regulation of cell communication | 19 | 42.73% | 2.75% |
| positive regulation of biological process | 27 | 82.73% | 1.46% | cellular response to peptide | 18.9 | 23.64% | 7.45% |
| regulation of cell communication | 26.7 | 64.55% | 2.13% | positive regulation of signaling | 18.9 | 42.73% | 2.74% |
| cellular response to organic substance | 26.5 | 56.36% | 2.59% | regulation of cell differentiation | 18.9 | 40.91% | 2.90% |
| cellular response to oxygen-containing compound | 26.2 | 42.73% | 4.03% | programmed cell death | 18.7 | 44.55% | 2.55% |
| positive regulation of cellular process | 24.1 | 77.27% | 1.49% | hormone secretion | 18.4 | 21.82% | 8.39% |
| response to peptide hormone | 24 | 28.18% | 7.79% | negative regulation of cellular process | 18.2 | 66.36% | 1.50% |
| glucose homeostasis | 23.2 | 23.64% | 10.83% | hormone transport | 18.1 | 21.82% | 8.14% |
| regulation of developmental process | 23.2 | 53.64% | 2.43% | small molecule metabolic process | 18.1 | 42.73% | 2.62% |
| carbohydrate homeostasis | 23.1 | 23.64% | 10.79% | regulation of molecular function | 18 | 53.64% | 1.93% |
| multicellular organismal process | 23 | 84.55% | 1.26% | Late onset | 17.9 | 23.38% | 41.86% |
| positive regulation of metabolic process | 22.4 | 63.64% | 1.85% | positive regulation of biosynthetic process | 17.9 | 44.55% | 2.45% |
| regulation of response to stimulus | 22.4 | 64.55% | 1.82% | regulation of phosphate metabolic process | 17.9 | 38.18% | 3.03% |
| Abnormal waist to hip ratio | 22 | 24.68% | 54.29% | regulation of phosphorus metabolic process | 17.9 | 38.18% | 3.03% |
| Increased waist to hip ratio | 22 | 24.68% | 54.29% | regulation of response to stress | 17.8 | 37.27% | 3.13% |
| response to insulin | 21.1 | 22.73% | 9.88% | regulation of hormone secretion | 17.6 | 20.00% | 9.32% |
| response to external stimulus | 20.9 | 54.55% | 2.15% | macromolecule localization | 17.4 | 53.64% | 1.89% |
| developmental process | 20.6 | 77.27% | 1.35% | regulation of intracellular signal transduction | 17.3 | 40.91% | 2.66% |
| cellular developmental process | 20.5 | 64.55% | 1.70% | cell surface receptor signaling pathway | 17.2 | 50.91% | 1.99% |
| regulation of cell population proliferation | 20.5 | 43.64% | 2.90% | positive regulation of cellular metabolic process | 17.2 | 54.55% | 1.83% |
| regulation of signal transduction | 20.5 | 55.45% | 2.07% | negative regulation of multicellular organismal process | 17 | 32.73% | 3.60% |
| regulation of apoptotic process | 20.4 | 40.91% | 3.17% | intracellular signal transduction | 16.9 | 49.09% | 2.05% |
| cellular response to nitrogen compound | 20.3 | 30.91% | 4.98% | cellular response to endogenous stimulus | 16.8 | 36.36% | 3.04% |
| cellular response to organonitrogen compound | 20.3 | 30.00% | 5.27% | anatomical structure development | 16.7 | 70.00% | 1.34% |
| regulation of cell death | 20.3 | 42.73% | 2.95% | organic substance transport | 16.7 | 49.09% | 2.03% |
| cell population proliferation | 20.2 | 46.36% | 2.60% | protein secretion | 16.7 | 21.82% | 7.08% |
| regulation of programmed cell death | 20.1 | 40.91% | 3.11% | establishment of protein localization to extracellular region | 16.6 | 21.82% | 7.06% |
| Insulin resistance | 20 | 29.87% | 29.87% | multicellular organismal homeostasis | 16.5 | 25.45% | 5.23% |
| cellular response to stimulus | 19.9 | 81.82% | 1.22% | positive regulation of cellular biosynthetic process | 16.5 | 42.73% | 2.39% |
| animal organ development | 19.8 | 59.09% | 1.85% | protein localization to extracellular region | 16.4 | 21.82% | 6.92% |
| cell differentiation | 19.8 | 63.64% | 1.68% | regulation of small molecule metabolic process | 16.4 | 21.82% | 6.92% |
| regulation of transport | 19.8 | 43.64% | 2.78% | regulation of protein localization | 16.3 | 30.00% | 3.91% |
| cell communication | 19.7 | 77.27% | 1.31% | multicellular organism development | 16.2 | 63.64% | 1.47% |
| response to hormone | 19.6 | 32.73% | 4.29% | negative regulation of biological process | 16.1 | 70.00% | 1.31% |
| ML Algorithm | Data | Marker Code | Marker Name | Importantance | Gene |
|---|---|---|---|---|---|
| DecisionTree | A | M313 | 209480_at | 8.54 | HLA-DQB1 |
| M399 | 212999_x_at | 6.83 | HLA-DQB1 | ||
| M398 | 212998_x_at | 5.98 | HLA-DQB1 | ||
| M710 | 238996_x_at | 5.98 | ALDOA | ||
| M370 | 211654_x_at | 5.12 | HLA-DQB1 | ||
| M417 | 214631_at | 5.12 | ZBTB33 | ||
| B | M148 | ILMN_1720311 | 13.07 | SLC25A46 | |
| M302 | ILMN_1790797 | 9.44 | VPS28 | ||
| M61 | ILMN_1672899 | 9.44 | POMC | ||
| M161 | ILMN_1726470 | 7.99 | OTUD5 | ||
| M41 | ILMN_1666192 | 7.99 | DCTN5 | ||
| M88 | ILMN_1684802 | 7.99 | TAF5 | ||
| RandomForest | A | M667 | 233510_s_at | 0.53 | PARVG |
| M710 | 238996_x_at | 0.41 | ALDOA | ||
| M313 | 209480_at | 0.40 | HLA-DQB1 | ||
| M546 | 223016_x_at | 0.25 | ZRANB2 | ||
| M203 | 205025_at | 0.19 | ZBTB48 | ||
| M141 | 202462_s_at | 0.18 | DDX46 | ||
| M399 | 212999_x_at | 0.16 | HLA-DQB1 | ||
| M636 | 230031_at | 0.15 | HSPA5 | ||
| M140 | 202455_at | 0.15 | HDAC5 | ||
| M80 | 1569150_x_at | 0.15 | PDLIM7 | ||
| B | M51 | ILMN_1670576 | 2.08 | IRF5 | |
| M41 | ILMN_1666192 | 1.97 | DCTN5 | ||
| M148 | ILMN_1720311 | 1.68 | SLC25A46 | ||
| M345 | ILMN_1813746 | 1.19 | CORO2A | ||
| M333 | ILMN_1806408 | 1.00 | ACADVL | ||
| M61 | ILMN_1672899 | 0.86 | POMC | ||
| M146 | ILMN_1718822 | 0.82 | STYXL1 | ||
| M239 | ILMN_1762095 | 0.81 | TMTC4 | ||
| M136 | ILMN_1709800 | 0.64 | POMZP3 | ||
| M265 | ILMN_1771697 | 0.52 | VRK3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsherbini, A.M.; Alsamman, A.M.; Elsherbiny, N.M.; El-Sherbiny, M.; Ahmed, R.; Ebrahim, H.A.; Bakkach, J. Decoding Diabetes Biomarkers and Related Molecular Mechanisms by Using Machine Learning, Text Mining, and Gene Expression Analysis. Int. J. Environ. Res. Public Health 2022, 19, 13890. https://doi.org/10.3390/ijerph192113890
Elsherbini AM, Alsamman AM, Elsherbiny NM, El-Sherbiny M, Ahmed R, Ebrahim HA, Bakkach J. Decoding Diabetes Biomarkers and Related Molecular Mechanisms by Using Machine Learning, Text Mining, and Gene Expression Analysis. International Journal of Environmental Research and Public Health. 2022; 19(21):13890. https://doi.org/10.3390/ijerph192113890
Chicago/Turabian StyleElsherbini, Amira M., Alsamman M. Alsamman, Nehal M. Elsherbiny, Mohamed El-Sherbiny, Rehab Ahmed, Hasnaa Ali Ebrahim, and Joaira Bakkach. 2022. "Decoding Diabetes Biomarkers and Related Molecular Mechanisms by Using Machine Learning, Text Mining, and Gene Expression Analysis" International Journal of Environmental Research and Public Health 19, no. 21: 13890. https://doi.org/10.3390/ijerph192113890
APA StyleElsherbini, A. M., Alsamman, A. M., Elsherbiny, N. M., El-Sherbiny, M., Ahmed, R., Ebrahim, H. A., & Bakkach, J. (2022). Decoding Diabetes Biomarkers and Related Molecular Mechanisms by Using Machine Learning, Text Mining, and Gene Expression Analysis. International Journal of Environmental Research and Public Health, 19(21), 13890. https://doi.org/10.3390/ijerph192113890