

Integrated Pangenome Analysis and Pharmacophore Modeling Revealed Potential Novel Inhibitors against Enterobacter xiangfangensis


 ,
, .jpg) ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieval and Pan Genome Analysis of Bacterial Proteome
2.2. Redundancy Analysis and Identification of Essential Proteins
2.3. Homology Analysis and Subcellular Localization
2.4. Identification of Virulent Proteins
2.5. Druggability Analysis and Drug Target Prioritization
2.6. Structure Prediction and Preparation
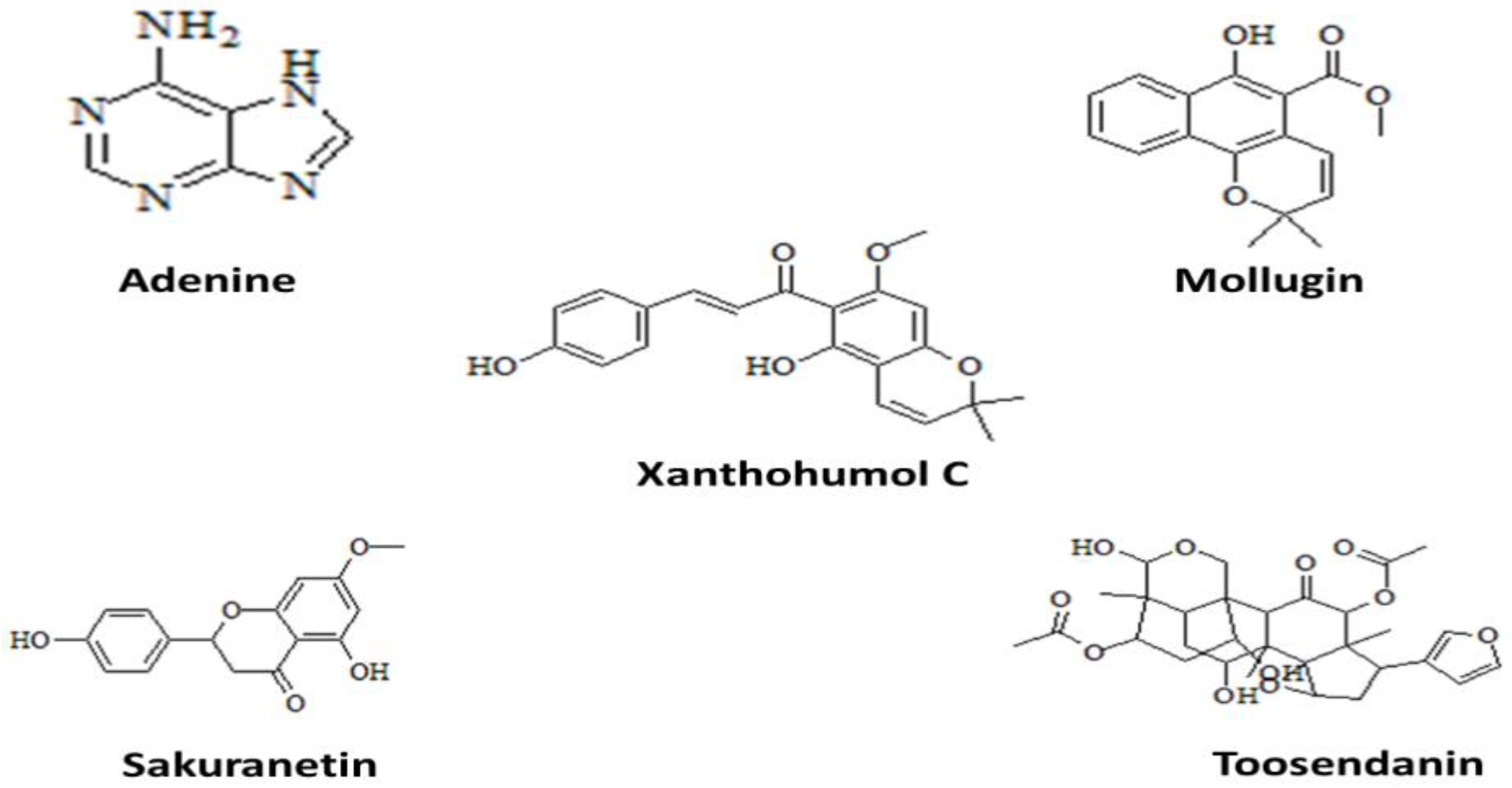
2.7. Ligands Retrieval
2.8. Molecular Docking and MD Simulation
2.9. Binding Free Energy Calculation
2.10. Physiochemical Profiling
3. Results
3.1. E. xiangfangensis Proteome Retrieval and Identification of Essential Nonhomologous Proteins
3.2. Subcellular Localization
3.3. Identification of Virulent Proteins and Druggability Analysis
3.4. Drug Target Prioritization
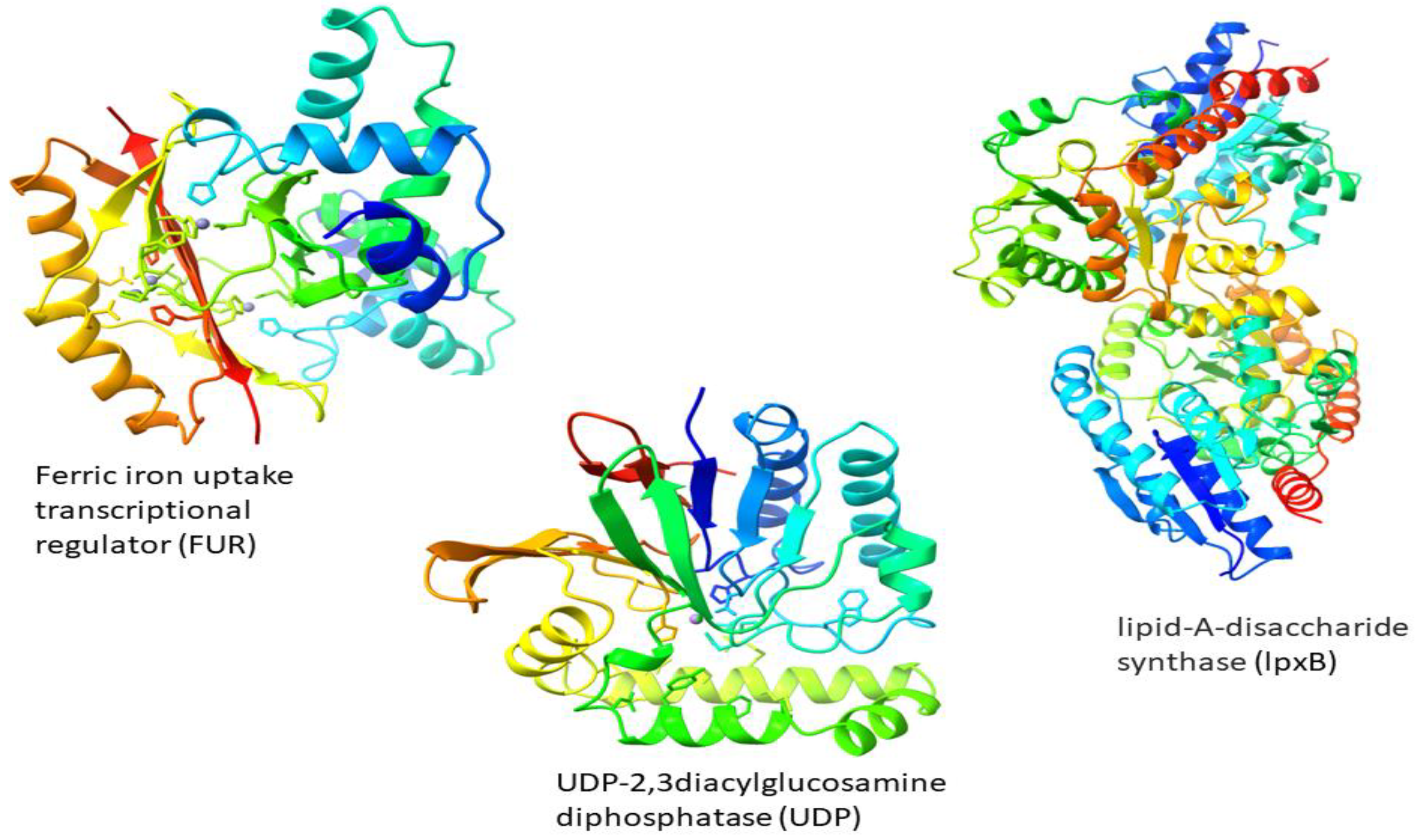
3.5. Structure Prediction
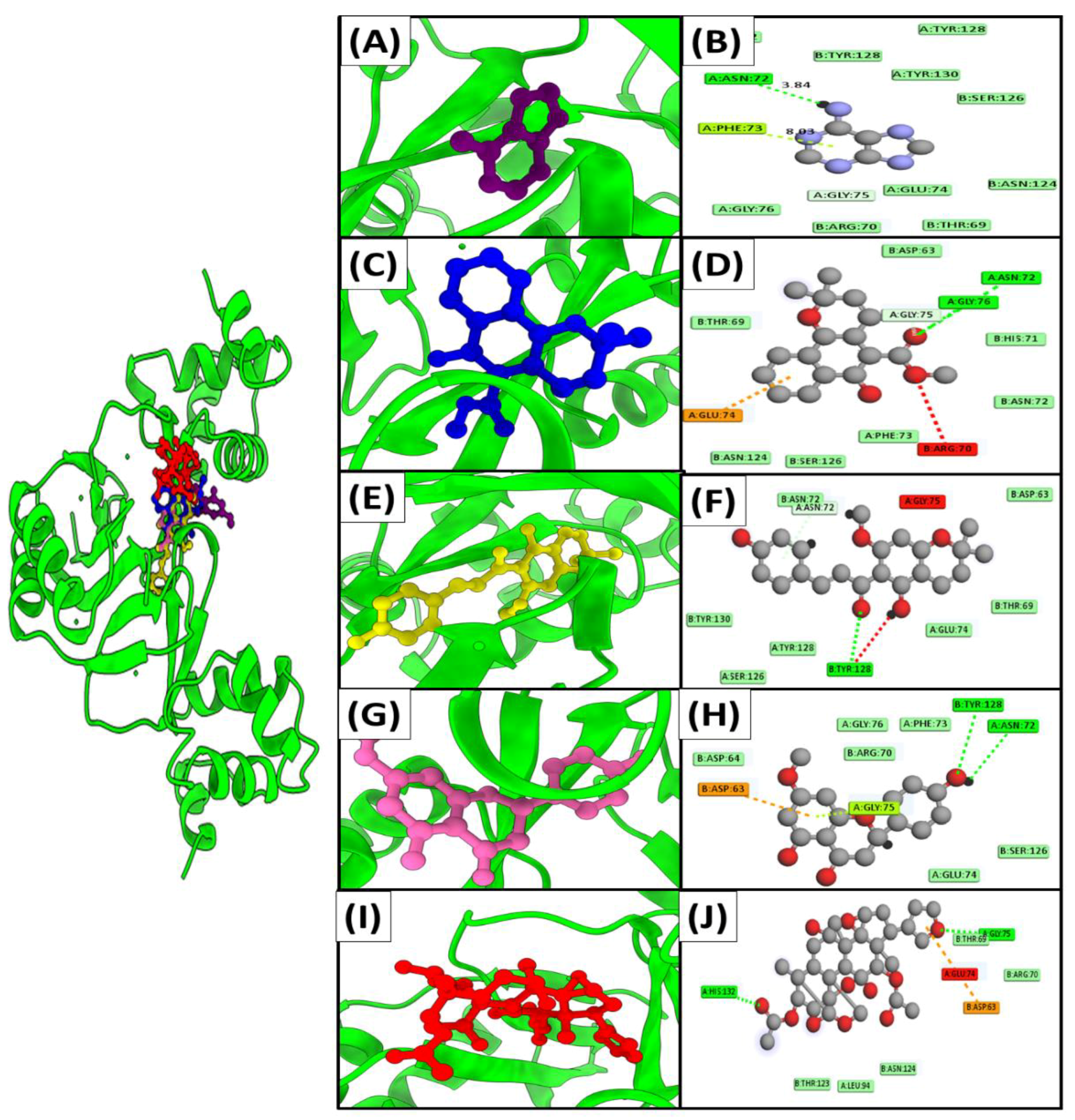
3.6. Molecular Docking
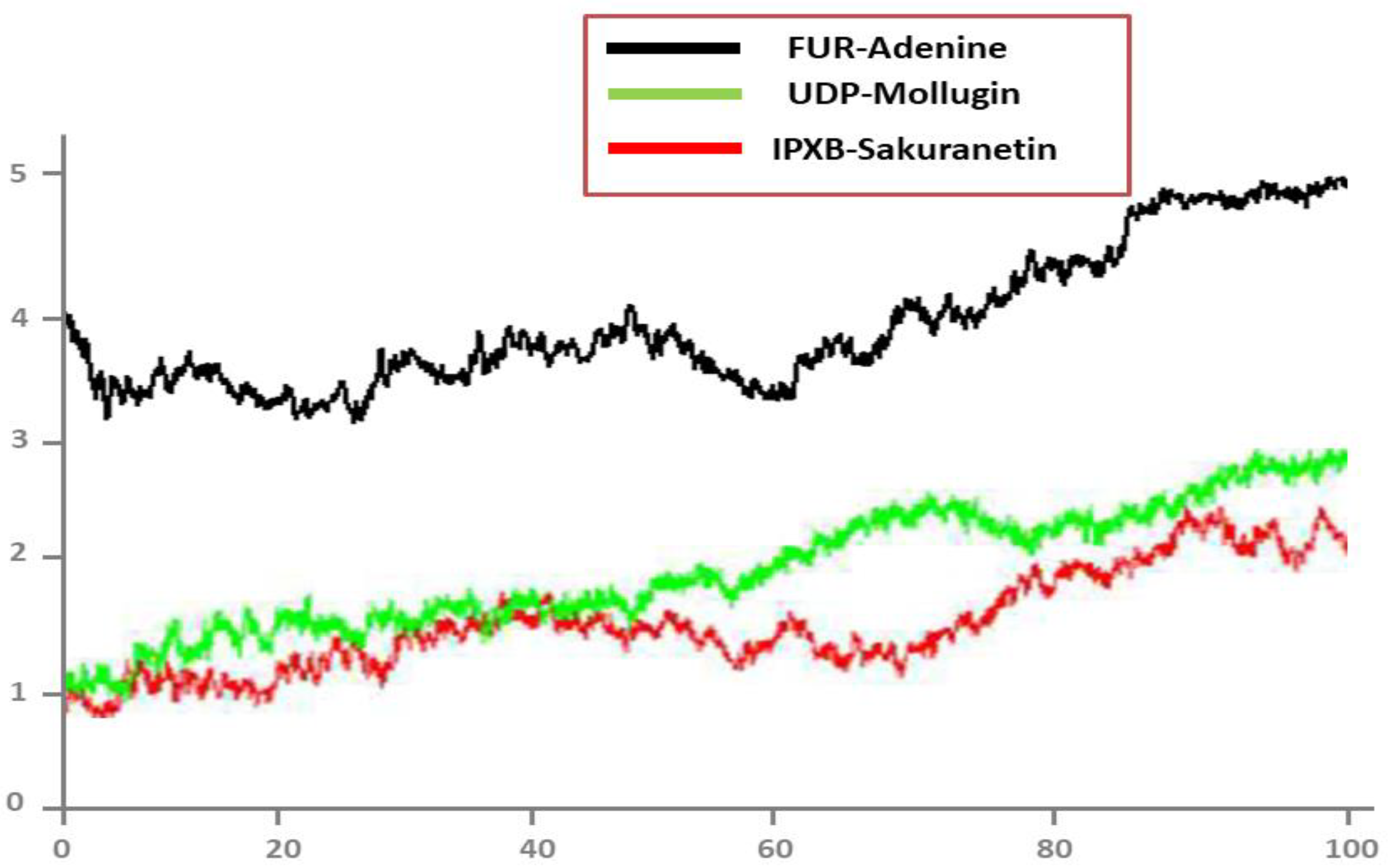
3.7. MD Simulation
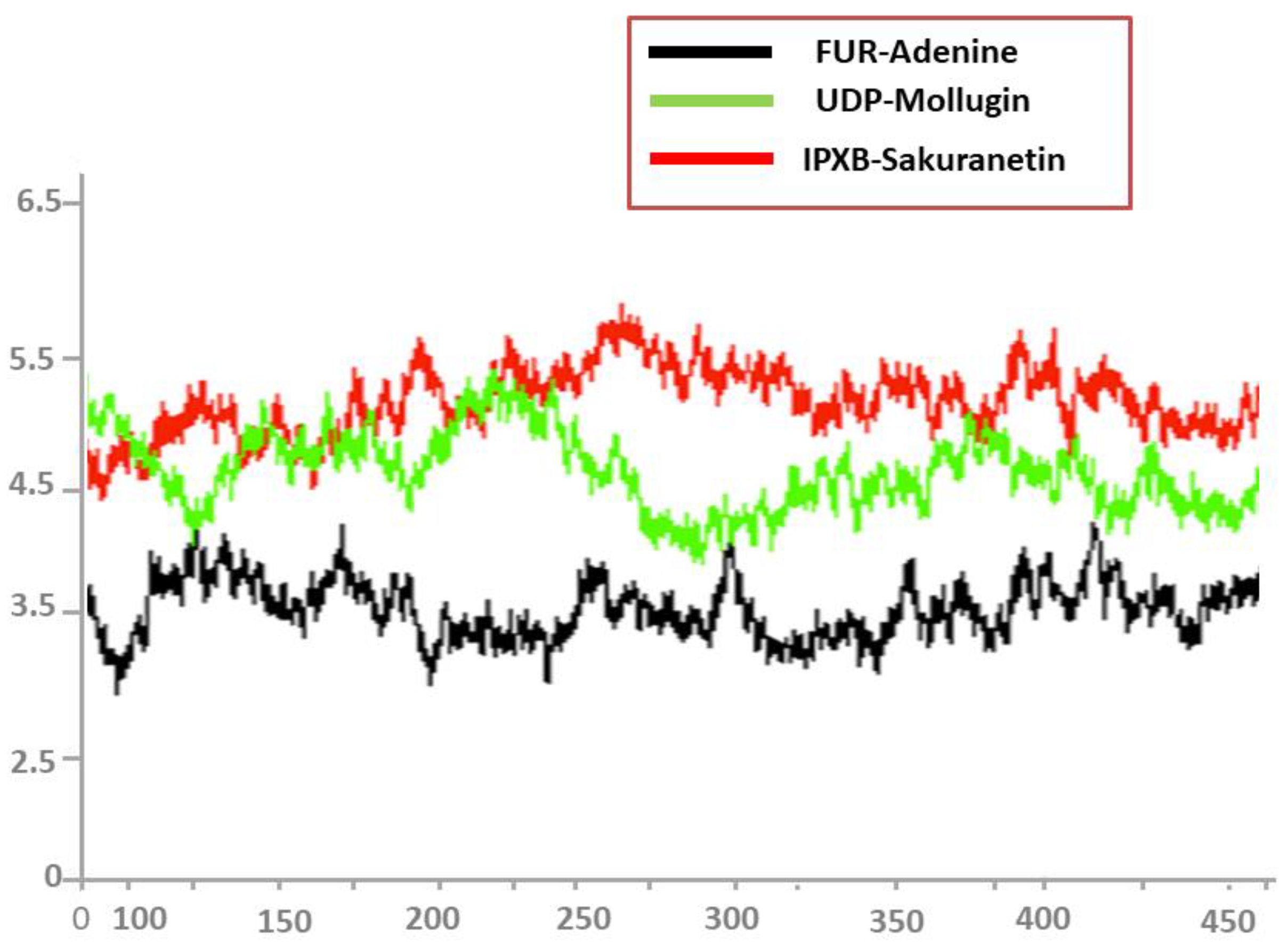
3.8. Root Mean Square Deviations (RMSD)
3.9. Root Mean Square Fluctuations (RMSF)
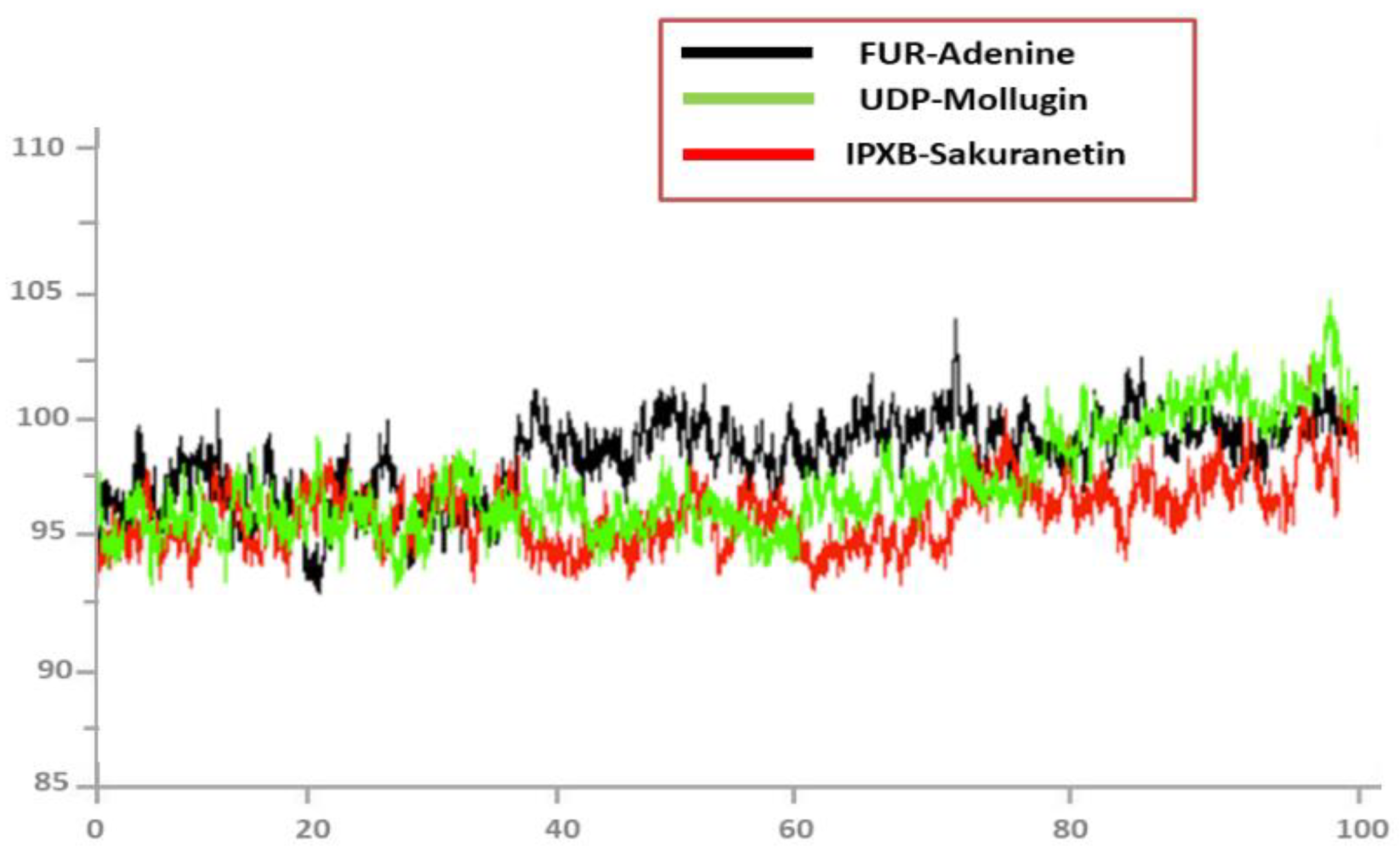
3.10. Radius of Gyration (RoG)
3.11. Binding Free Energy Calculations
3.12. Drug Scan/ADMET
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ramirez, D.; Giron, M. Enterobacter infections. In StatPearls; StatPearls Publishing: Lake Worth, FL, USA, 2021. [Google Scholar]
- Davin-Regli, A.; Pagès, J.-M. Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front. Microbiol. 2015, 6, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, W.E., Jr.; Sanders, C.C. Enterobacter spp.: Pathogens poised to flourish at the turn of the century. Clin. Microbiol. Rev. 1997, 10, 220–241. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Series Introduction: Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamptey, R.N.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef] [PubMed]
- Ziukelis, E.T.; Mak, E.; Dounavi, M.-E.; Su, L.; O’Brien, J. Fractal dimension of the brain in neurodegenerative disease and dementia: A systematic review. Ageing Res. Rev. 2022, 79, 101651. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scorza, F.A.; Guimarães-Marques, M.; Nejm, M.; de Almeida, A.C.G.; Scorza, C.A.; Fiorini, A.C.; Finsterer, J. Sudden unexpected death in Parkinson’s disease: Insights from clinical practice. Clinics 2022, 77, 100001. [Google Scholar] [CrossRef]
- Basharat, Z.; Jahanzaib, M.; Rahman, N. Therapeutic target identification via differential genome analysis of antibiotic resistant Shigella sonnei and inhibitor evaluation against a selected drug target. Infect. Genet. Evol. 2021, 94, 105004. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative diseases–is metabolic deficiency the root cause? Front. Neurosci. 2020, 14, 213. [Google Scholar] [CrossRef] [Green Version]
- Aslam, S.; Mehmood, M.A.; Rahman, M.-u.; Noor, F.; Ahmad, N. Bioinformatics-assisted multiomics approaches to improve the agronomic traits in cotton. In Bioinformatics in Agriculture; Elsevier: Amsterdam, The Netherlands, 2022; pp. 233–251. [Google Scholar]
- Noor, F.; Ahmad, S.; Saleem, M.; Alshaya, H.; Qasim, M.; Rehman, A.; Ehsan, H.; Talib, N.; Saleem, H.; Bin Jardan, Y.A. Designing a multi-epitope vaccine against Chlamydia pneumoniae by integrating the core proteomics, subtractive proteomics and reverse vaccinology-based immunoinformatics approaches. Comput. Biol. Med. 2022, 145, 105507. [Google Scholar] [CrossRef]
- Noor, F.; Ashfaq, U.A.; Javed, M.R.; Saleem, M.H.; Ahmad, A.; Aslam, M.F.; Aslam, S. Comprehensive computational analysis reveals human respiratory syncytial virus encoded microRNA and host specific target genes associated with antiviral immune responses and protein binding. J. King Saud Univ.-Sci. 2021, 33, 101562. [Google Scholar] [CrossRef]
- Noor, F.; Noor, A.; Ishaq, A.R.; Farzeen, I.; Saleem, M.H.; Ghaffar, K.; Aslam, M.F.; Aslam, S.; Chen, J.-T. Recent advances in diagnostic and therapeutic approaches for breast cancer: A comprehensive review. Curr. Pharm. Des. 2021, 27, 2344–2365. [Google Scholar] [CrossRef] [PubMed]
- Choonara, Y.E.; Pillay, V.; Du Toit, L.C.; Modi, G.; Naidoo, D.; Ndesendo, V.M.; Sibambo, S.R. Trends in the molecular pathogenesis and clinical therapeutics of common neurodegenerative disorders. Int. J. Mol. Sci. 2009, 10, 2510–2557. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M. New hopes and challenges for treatment of neurodegenerative disorders: Great opportunities for young neuroscientists. Basic Clin. Neurosci. 2013, 4, 3. [Google Scholar]
- Almatroudi, A. Non-Coding RNAs in Tuberculosis Epidemiology: Platforms and Approaches for Investigating the Genome’s Dark Matter. Int. J. Mol. Sci. 2022, 23, 4430. [Google Scholar] [CrossRef]
- Das, T.; Das, T.K.; Khodarkovskaya, A.; Dash, S. Non-coding RNAs and their bioengineering applications for neurological diseases. Bioengineered 2021, 12, 11675–11698. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Zhu, X.; Khan, M.S.; Xing, F.; Chen, L.-L. Pan-genome: A promising resource for noncoding RNA discovery in plants. Plant Genome 2020, 13, e20046. [Google Scholar]
- Koch, A.; Cox, H.; Mizrahi, V. Drug-resistant tuberculosis: Challenges and opportunities for diagnosis and treatment. Curr. Opin. Pharmacol. 2018, 42, 7–15. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Mehler, M.F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 2012, 13, 528–541. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.; Wang, X.; Ahmad, S.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Alrumaihi, F.; Qasim, M.; Hashem, A.; Al-Hazzani, A.A. In Silico Core Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pyogenes. Int. J. Environ. Res. Public Health 2021, 18, 11355. [Google Scholar] [CrossRef]
- Salvatori, B.; Biscarini, S.; Morlando, M. Non-coding RNAs in nervous system development and disease. Front. Cell Dev. Biol. 2020, 8, 273. [Google Scholar] [CrossRef] [PubMed]
- Dahariya, S.; Paddibhatla, I.; Kumar, S.; Raghuwanshi, S.; Pallepati, A.; Gutti, R.K. Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol. Immunol. 2019, 112, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Slaby, O.; Calin, G.A. Non-Coding RNAs in Colorectal Cancer; Springer: Berlin, Germany, 2016; Volume 937. [Google Scholar]
- Nelson, W.C.; Stegen, J.C. The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front. Microbiol. 2015, 6, 713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memon, D.; Bi, J.; Miller, C.J. In silico prediction of housekeeping long intergenic non-coding RNAs reveals HKlincR1 as an essential player in lung cancer cell survival. Sci. Rep. 2019, 9, 7372. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Lin, Y.; Wu, J. Long non-coding RNA expression profiling of mouse testis during postnatal development. PLoS ONE 2013, 8, e75750. [Google Scholar] [CrossRef] [PubMed]
- Guglas, K.; Bogaczyńska, M.; Kolenda, T.; Ryś, M.; Teresiak, A.; Bliźniak, R.; Łasińska, I.; Mackiewicz, J.; Lamperska, K. lncRNA in HNSCC: Challenges and potential. Contemp. Oncol. Współczesna Onkol. 2017, 21, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, D.E.; Martin, M.M.; Feldman, D.S.; Terry, A.V., Jr.; Nuovo, G.J.; Elton, T.S. Experimental validation of miRNA targets. Methods 2008, 44, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Bavelloni, A.; Ramazzotti, G.; Poli, A.; Piazzi, M.; Focaccia, E.; Blalock, W.; Faenza, I. MiRNA-210: A current overview. Anticancer Res. 2017, 37, 6511–6521. [Google Scholar]
- Noor, F.; Saleem, M.H.; Aslam, M.F.; Ahmad, A.; Aslam, S. Construction of miRNA-mRNA network for the identification of key biological markers and their associated pathways in IgA nephropathy by employing the integrated bioinformatics analysis. Saudi J. Biol. Sci. 2021, 28, 4938–4945. [Google Scholar] [CrossRef]
- Sufyan, M.; Ashfaq, U.A.; Ahmad, S.; Noor, F.; Saleem, M.H.; Aslam, M.F.; El-Serehy, H.A.; Aslam, S. Identifying key genes and screening therapeutic agents associated with diabetes mellitus and HCV-related hepatocellular carcinoma by bioinformatics analysis. Saudi J. Biol. Sci. 2021, 28, 5518–5525. [Google Scholar] [CrossRef]
- Noor, F.; Saleem, M.H.; Javed, M.R.; Chen, J.-T.; Ashfaq, U.A.; Okla, M.K.; Abdel-Maksoud, M.A.; Alwasel, Y.A.; Al-Qahtani, W.H.; Alshaya, H.; et al. Comprehensive computational analysis reveals H5N1 influenza virus-encoded miRNAs and host-specific targets associated with antiviral immune responses and protein binding. PLoS ONE 2022, 17, e0263901. [Google Scholar] [CrossRef] [PubMed]
- Gan, E.-S.; Huang, J.; Ito, T. Functional roles of histone modification, chromatin remodeling and microRNAs in Arabidopsis flower development. Int. Rev. Cell Mol. Biol. 2013, 305, 115–161. [Google Scholar] [PubMed]
- Beermann, J.; Piccoli, M.-T.; Viereck, J.; Thum, T. Non-coding RNAs in development and disease: Background, mechanisms, and therapeutic approaches. Physiol. Rev. 2016, 96, 4. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Curtis, H.J.; Sibley, C.R.; Wood, M.J. Mirtrons, an emerging class of atypical miRNA. Wiley Interdiscip. Rev. RNA 2012, 3, 617–632. [Google Scholar] [CrossRef]
- Juźwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef]
- Lang, M.-F.; Shi, Y. Dynamic roles of microRNAs in neurogenesis. Front. Neurosci. 2012, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α–PU. 1 pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef]
- Coolen, M.; Katz, S.; Bally-Cuif, L. miR-9: A versatile regulator of neurogenesis. Front. Cell. Neurosci. 2013, 7, 220. [Google Scholar] [CrossRef] [Green Version]
- Pathania, M.; Torres-Reveron, J.; Yan, L.; Kimura, T.; Lin, T.V.; Gordon, V.; Teng, Z.-Q.; Zhao, X.; Fulga, T.A.; Van Vactor, D. miR-132 enhances dendritic morphogenesis, spine density, synaptic integration, and survival of newborn olfactory bulb neurons. PLoS ONE 2012, 7, e38174. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Reczko, M.; Maragkakis, M.; Dalamagas, T.M.; Hatzigeorgiou, A.G. DIANA-LncBase: Experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013, 41, D239–D245. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, G.-Y. Novel human lncRNA–disease association inference based on lncRNA expression profiles. Bioinformatics 2013, 29, 2617–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferre, F.; Colantoni, A.; Helmer-Citterich, M. Revealing protein–lncRNA interaction. Brief. Bioinform. 2016, 17, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Tanigawa, A.; Naganuma, T.; Ohkawa, Y.; Souquere, S.; Pierron, G.; Hirose, T. SWI/SNF chromatin-remodeling complexes function in noncoding RNA-dependent assembly of nuclear bodies. Proc. Natl. Acad. Sci. USA 2015, 112, 4304–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinodoz, S.; Guttman, M. Long noncoding RNAs: An emerging link between gene regulation and nuclear organization. Trends Cell Biol. 2014, 24, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.A.; Khan, F.B.; Safdari, H.A.; Almatroudi, A.; Alzohairy, M.A.; Safdari, M.; Amirizadeh, M.; Rehman, S.; Equbal, M.J.; Hoque, M. Prospective therapeutic potential of Tanshinone IIA: An updated overview. Pharmacol. Res. 2021, 164, 105364. [Google Scholar] [CrossRef]
- Parasramka, M.A.; Maji, S.; Matsuda, A.; Yan, I.K.; Patel, T. Long non-coding RNAs as novel targets for therapy in hepatocellular carcinoma. Pharmacol. Ther. 2016, 161, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E. Translation of circRNAs. Mol. Cell 2017, 66, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Basharat, Z.; Jahanzaib, M.; Yasmin, A.; Khan, I.A. Pan-genomics, drug candidate mining and ADMET profiling of natural product inhibitors screened against Yersinia pseudotuberculosis. Genomics 2021, 113, 238–244. [Google Scholar] [CrossRef]
- Saleem, H.; Ashfaq, U.A.; Nadeem, H.; Zubair, M.; Siddique, M.H.; Rasul, I. Subtractive genomics and molecular docking approach to identify drug targets against Stenotrophomonas maltophilia. PLoS ONE 2021, 16, e0261111. [Google Scholar] [CrossRef]
- Dar, H.A.; Zaheer, T.; Ullah, N.; Bakhtiar, S.M.; Zhang, T.; Yasir, M.; Azhar, E.I.; Ali, A. Pangenome Analysis of Mycobacterium tuberculosis Reveals Core-Drug Targets and Screening of Promising Lead Compounds for Drug Discovery. Antibiotics 2020, 9, 819. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, N.A.; Bakhtiar, S.M.; Faheem, M.; Shah, M.; Bari, A.; Mahmood, H.M.; Sohaib, M.; Mothana, R.A.; Ullah, R.; Jamal, S.B. Genome-based drug target identification in human pathogen Streptococcus gallolyticus. Front. Genet. 2021, 12, 564056. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Harries, L.W. Circular RNAs (circRNAs) in health and disease. Genes 2017, 8, 353. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Weng, X.; Zhao, Y.; Chen, W.; Gan, T.; Xu, D. Circular RNAs in cardiovascular disease: An overview. BioMed Res. Int. 2017, 2017, 5135781. [Google Scholar] [CrossRef] [Green Version]
- Batool, S.; Javed, M.R.; Aslam, S.; Noor, F.; Javed, H.M.F.; Seemab, R.; Rehman, A.; Aslam, M.F.; Paray, B.A.; Gulnaz, A.J.P. Network Pharmacology and Bioinformatics Approach Reveals the Multi-Target Pharmacological Mechanism of Fumaria indica in the Treatment of Liver Cancer. Pharmaceuticals 2022, 15, 654. [Google Scholar] [CrossRef]
- Noor, F.; Rehman, A.; Ashfaq, U.A.; Saleem, M.H.; Okla, M.K.; Al-Hashimi, A.; AbdElgawad, H.; Aslam, S.J.P. Integrating Network Pharmacology and Molecular Docking Approaches to Decipher the Multi-Target Pharmacological Mechanism of Abrus precatorius L. Acting on Diabetes. Pharmaceuticals 2022, 15, 414. [Google Scholar] [CrossRef]
- Noor, F.; Tahir ul Qamar, M.; Ashfaq, U.A.; Albutti, A.; Alwashmi, A.S.; Aljasir, M.A. Network Pharmacology Approach for Medicinal Plants: Review and Assessment. Pharmaceuticals 2022, 15, 572. [Google Scholar] [CrossRef]
- Ebbesen, K.K.; Kjems, J.; Hansen, T.B. Circular RNAs: Identification, biogenesis and function. Biochim. Biophys. Acta BBA-Gene Regul. Mech. 2016, 1859, 163–168. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Subcellular Localization | Transmembrane Helices | Molecular Weight | Stability | Molecular Function | Biological Processes |
|---|---|---|---|---|---|---|
| Ferric iron uptake transcriptional regulator (FUR) | Cytoplasm | 0 | 16,765.81 | Stable | DNA-binding transcription factor activity | regulation of transcription, DNA-templated |
| UDP-2,3diacylglucosamine diphosphatase (UDP) | Cytoplasm | 0 | 26,832.02 | Stable | pyrophosphatase activity hydrolase activity | lipid A biosynthetic process |
| lipid-A-disaccharide synthase (lpxB) | Cytoplasm | 0 | 42,472.56 | Stable | lipid-A-disaccharide synthase activity | lipid A biosynthetic process |
| Scores | FUR Protein | UDP Protein | lpxB Protein |
|---|---|---|---|
| C-score | −6.02 | −4.98 | −7.87 |
| Estimated TM-score | 0.91 ± 0.05 | 0.85 ± 0.09 | 0.74 ± 0.08 |
| ProSA | |||
| Z Score | −7.65 | −8.35 | −6.98 |
| Verify 3D | |||
| Compatibility Score | 81.71 | 83.89 | 80.03 |
| Errat | |||
| Quality Factor | 91.76 | 87.56 | 90.67 |
| Ramachandran plot (%) | |||
| Core | 90.2% | 83.7% | 88.7% |
| Allowed | 6.6% | 12.8% | 7.9% |
| General | 2.0% | 1.4% | 2.9% |
| Disallowed | 1.9% | 1.5% | 1.8% |
| Compounds Name and ID | FUR Protein | UDP Protein | lpxB Protein | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Binding Affinity | Inhibition Constant | RMSD | Interacting Residues | Binding Affinity | Inhibition Constant | RMSD | Interacting Residues | Binding Affinity | Inhibition Constant | RMSD | Interacting Residues | |
| Adenine (190) | −18.7 | 67.1 μM | 0.9 | Asn A72,Phe A73,Gly A75,Glu A74 | −11.6 | 58.7 μM | 2.5 | Cys A119,His A195,Tyr A125,Lys A167,Asp A122,Met A172 | −15.3 | 69.9 μM | 1.8 | Phe A153,Trp B301,Lys B304 |
| Mollugin (124219) | −16.2 | 72.2 μM | 1.2 | Glu A74,Arg B70,Gly A76,Asn A72,Gly A75 | −19.8 | 75.2 μM | 0.7 | Tyr A125,Cys A119,Met A172,His A197 | −18.8 | 89.6 μM | 1.1 | Trp B301,Phe A153,Lys B 304 |
| xanthohumol C (10338075) | −14.5 | 85.2 μM | 1.8 | Tyr B128,Asn B72,Gly A75, | −15.3 | 80.1 μM | 1.5 | Ala A153,Ala A45,Met A156 | −16.2 | 72.7 μM | 0.8 | Leu A147,Leu B314,Phe A153,Trp B301 |
| Sakuranetin (73571) | −13.6 | 76.4 μM | 0.8 | Tyr B128,Asn A72,Asp B63 | −14.9 | 90.3 μM | 2.1 | Ser A160,Asn A79,Phe A 128,Ala A163,Asn A164 | −19.3 | 80.2 μM | 2.3 | Phe A153,Lys B304,Leu B317,Lys B308 |
| Toosendanin (115060) | −13.1 | 93.1 μM | 2.0 | His A132,Thr B69,Gly A75,GluA74,Asp B63 | −17.6 | 63.9 μM | 0.9 | Asn A164,Arg A80,Asn A79,His A10 | −14.3 | 70.4 μM | 2.9 | Lys B308,Arg A156,Ser A124,Trp B301 |
| Energy Component | Adenine | Mollugin | Xanthohumol C | Sakuranetin | Toosendanin |
|---|---|---|---|---|---|
| Van der Waals | −45.61 | −34.06 | −39.19 | −42.12 | −44.71 |
| Electrostatic | −41.95 | −26.23 | −37.69 | −34.69 | −33.96 |
| Polar solvation | 59.79 | 45.10 | 52.45 | 55.02 | 65.08 |
| Nonpolar solvation | −4.40 | −6.90 | −5.32 | −4.49 | −7.70 |
| Net gas phase | −78.23 | −70.79 | −61.12 | −45.05 | −59.45 |
| Net solvation | 60.28 | 55.17 | 46.41 | 61.77 | 45.31 |
| Net complex energy | −35.52 | −50.18 | −45.41 | −42.21 | −50.45 |
| Ligands | Molecular Weight | Molecular Formula | Hydrogen Bond Donor | Hydrogen Bond Acceptor | XLogP3 | Heavy Atom Count |
|---|---|---|---|---|---|---|
| Adenine | 135.13 | C5H5N5 | 2 | 4 | −0.1 | 10 |
| Mollugin | 284.31 | C17H16O4 | 1 | 4 | 4.1 | 21 |
| xanthohumol C | 352.4 | C21H20O5 | 2 | 5 | 4.4 | 26 |
| Sakuranetin | 286.28 | C16H14O5 | 2 | 5 | 2.7 | 21 |
| Toosendanin | 574.6 | C30H38O11 | 3 | 10 | 0.7 | 41 |
| Compounds | Adenine | Mollugin | Xanthohumol C | Sakuranetin | Toosendanin |
|---|---|---|---|---|---|
| Absorption/Distribution | |||||
| Blood–Brain Barrier | No | No | No | No | No |
| Log S | −410 | −3.70 | −4.12 | −4.76 | −4.94 |
| GI Absorption | High | Low | High | High | Low |
| Caco-2 permeability | −5.18 | −8.98 | −6.71 | −6.54 | −7.72 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.17 |
| Metabolism | |||||
| P-gp substrate | No | No | Yes | No | No |
| CYP1A2 inhibitor | No | Yes | No | Yes | Yes |
| CYP2C19 inhibitor | No | No | Yes | Yes | Yes |
| CYP2C9 inhibitor | No | Yes | No | No | Yes |
| CYP2D6 inhibitor | No | Yes | Yes | No | No |
| CYP3A4 inhibitor | No | Yes | Yes | Yes | Yes |
| Toxicity | |||||
| AMES Toxicity | Nill | Nill | Nill | Nill | Nill |
| Carcinogenicity | None | None | None | None | None |
| Immunogenicity | NT | NT | NT | NT | NT |
| Acute Oral Toxicity | NT | NT | NT | NT | NT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almuhayawi, M.S.; Al Jaouni, S.K.; Selim, S.; Alkhalifah, D.H.M.; Marc, R.A.; Aslam, S.; Poczai, P. Integrated Pangenome Analysis and Pharmacophore Modeling Revealed Potential Novel Inhibitors against Enterobacter xiangfangensis. Int. J. Environ. Res. Public Health 2022, 19, 14812. https://doi.org/10.3390/ijerph192214812
Almuhayawi MS, Al Jaouni SK, Selim S, Alkhalifah DHM, Marc RA, Aslam S, Poczai P. Integrated Pangenome Analysis and Pharmacophore Modeling Revealed Potential Novel Inhibitors against Enterobacter xiangfangensis. International Journal of Environmental Research and Public Health. 2022; 19(22):14812. https://doi.org/10.3390/ijerph192214812
Chicago/Turabian StyleAlmuhayawi, Mohammed S., Soad K. Al Jaouni, Samy Selim, Dalal Hussien M. Alkhalifah, Romina Alina Marc, Sidra Aslam, and Peter Poczai. 2022. "Integrated Pangenome Analysis and Pharmacophore Modeling Revealed Potential Novel Inhibitors against Enterobacter xiangfangensis" International Journal of Environmental Research and Public Health 19, no. 22: 14812. https://doi.org/10.3390/ijerph192214812
APA StyleAlmuhayawi, M. S., Al Jaouni, S. K., Selim, S., Alkhalifah, D. H. M., Marc, R. A., Aslam, S., & Poczai, P. (2022). Integrated Pangenome Analysis and Pharmacophore Modeling Revealed Potential Novel Inhibitors against Enterobacter xiangfangensis. International Journal of Environmental Research and Public Health, 19(22), 14812. https://doi.org/10.3390/ijerph192214812