






Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes


Abstract
:1. Introduction
2. Materials and Methods
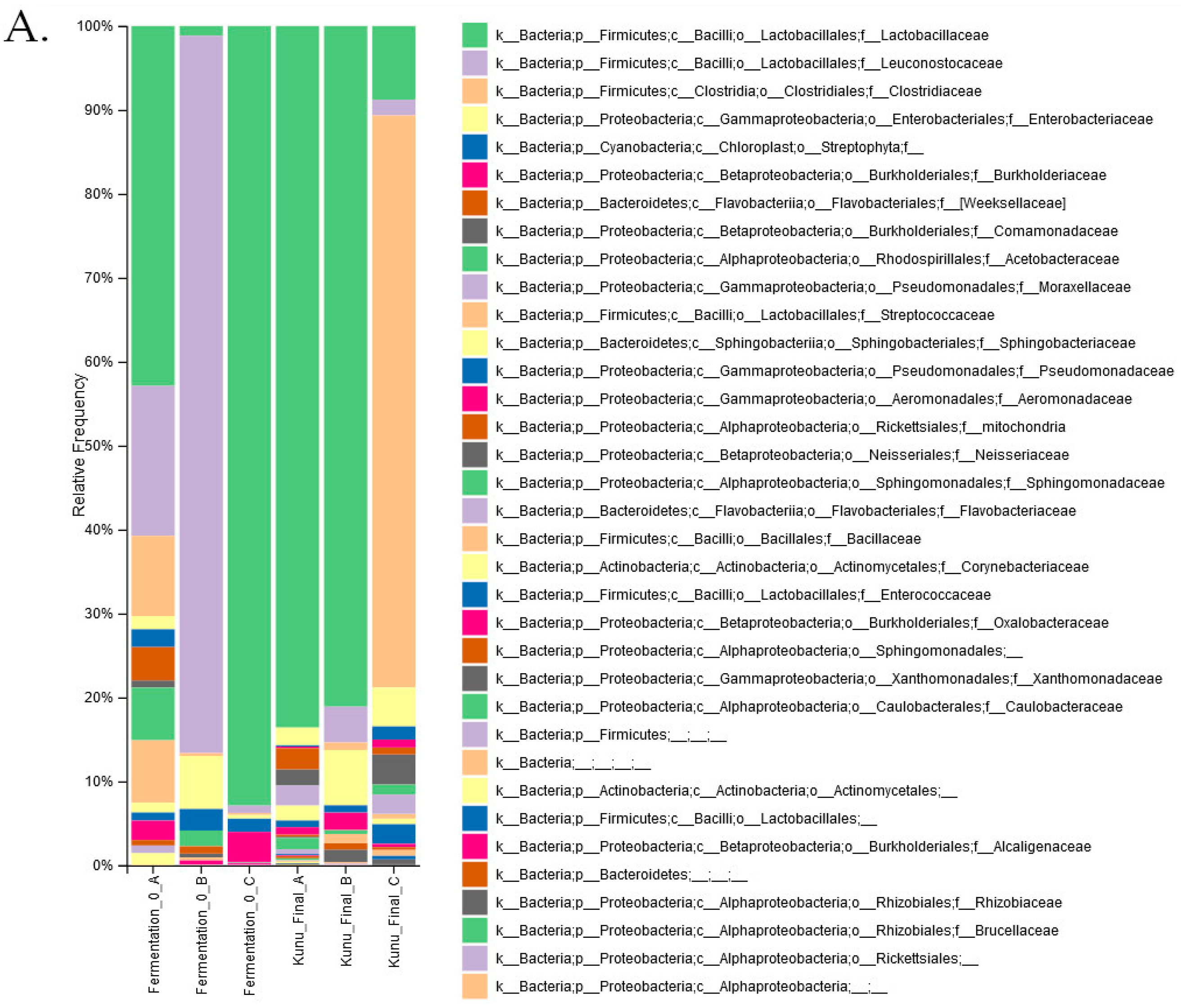
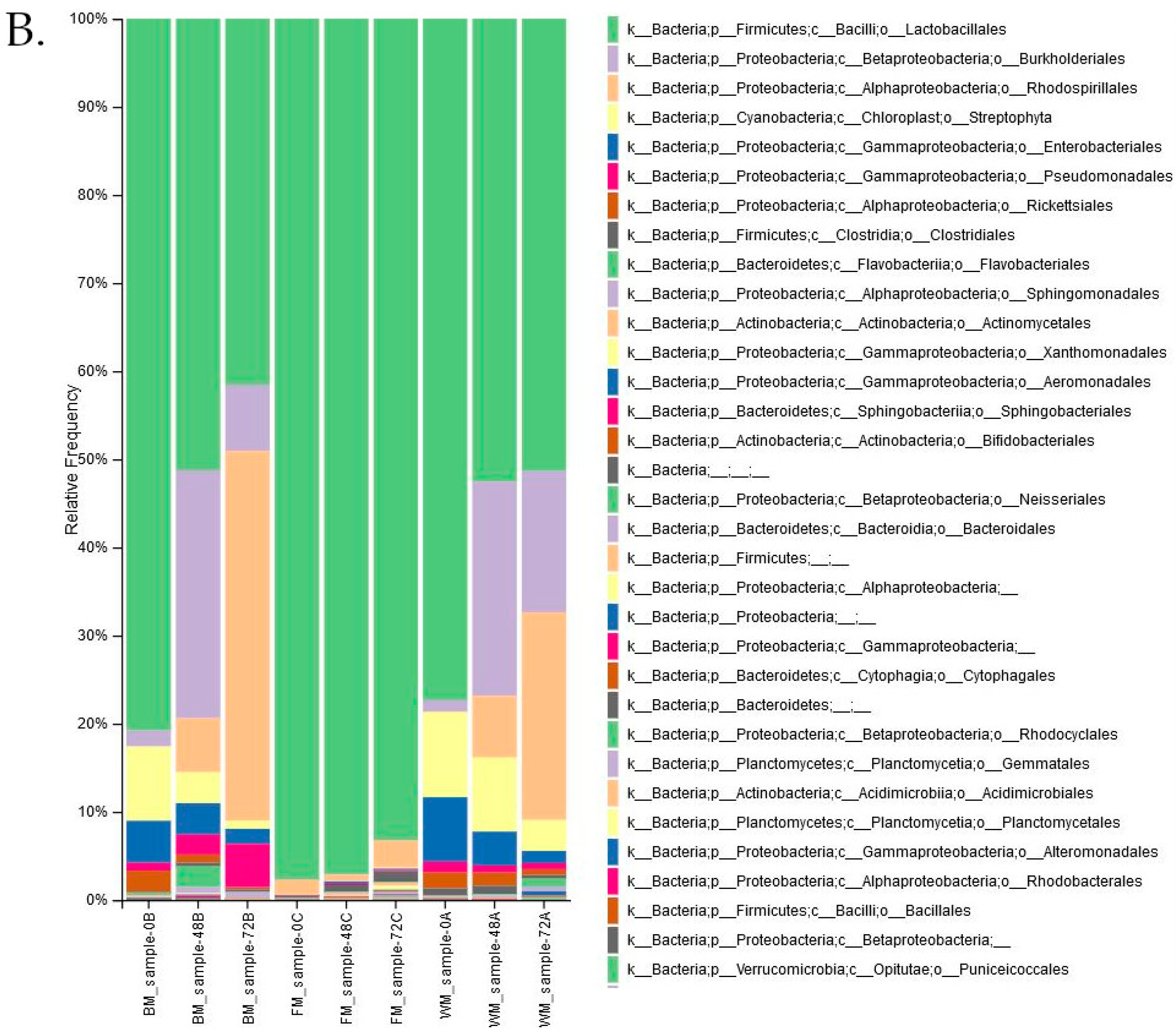
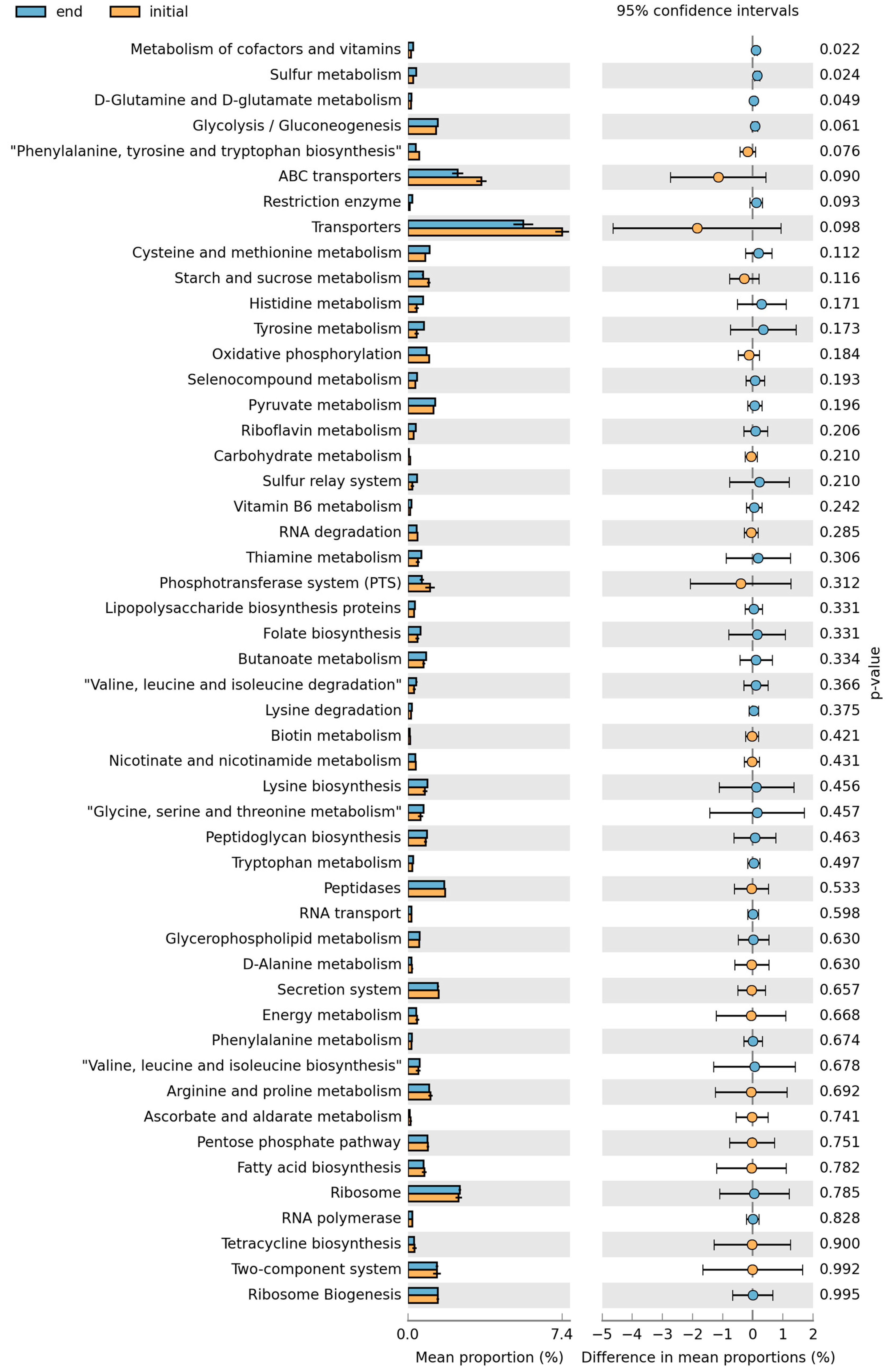
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sivamaruthi, B.; Kesika, P.; Chaiyasut, C. Thai Fermented Foods as a Versatile Source of Bioactive Microorganisms—A Comprehensive Review. Sci. Pharm. 2018, 86, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves-López, C.; Serio, A.; Grande-Tovar, C.D.; Cuervo-Mulet, R.; Delgado-Ospina, J.; Paparella, A. Traditional Fermented Foods and Beverages from a Microbiological and Nutritional Perspective: The Colombian Heritage: Colombian Fermented Foods and Beverages. Compr. Rev. Food Sci. Food Saf. 2014, 13, 1031–1048. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Tayie, F.A.K.; Young, M.F.; Rocheford, T.; White, W.S. Retention of Provitamin A Carotenoids in High β-Carotene Maize (Zea Mays) During Traditional African Household Processing. J. Agric. Food Chem. 2007, 55, 10744–10750. [Google Scholar] [CrossRef] [PubMed]
- Blandino, A.; Al-Aseeri, M.E.; Pandiella, S.S.; Cantero, D.; Webb, C. Cereal-Based Fermented Foods and Beverages. Food Res. Int. 2003, 36, 527–543. [Google Scholar] [CrossRef]
- Messia, M.C.; Reale, A.; Maiuro, L.; Candigliota, T.; Sorrentino, E.; Marconi, E. Effects of Pre-Fermented Wheat Bran on Dough and Bread Characteristics. J. Cereal Sci. 2016, 69, 138–144. [Google Scholar] [CrossRef]
- Pranoto, Y.; Anggrahini, S.; Efendi, Z. Effect of Natural and Lactobacillus Plantarum Fermentation on In-Vitro Protein and Starch Digestibilities of Sorghum Flour. Food Biosci. 2013, 2, 46–52. [Google Scholar] [CrossRef]
- Tangyu, M.; Muller, J.; Bolten, C.J.; Wittmann, C. Fermentation of Plant-Based Milk Alternatives for Improved Flavour and Nutritional Value. Appl. Microbiol. Biotechnol. 2019, 103, 9263–9275. [Google Scholar] [CrossRef] [Green Version]
- Tamene, A.; Baye, K.; Kariluoto, S.; Edelmann, M.; Bationo, F.; Leconte, N.; Humblot, C. Lactobacillus Plantarum P2R3FA Isolated from Traditional Cereal-Based Fermented Food Increase Folate Status in Deficient Rats. Nutrients 2019, 11, 2819. [Google Scholar] [CrossRef] [Green Version]
- Steinkraus, K.H. Classification of Fermented Foods: Worldwide Review of Household Fermentation Techniques. Food Control 1997, 8, 311–317. [Google Scholar] [CrossRef]
- Poutanen, K.S.; Kårlund, A.O.; Gómez-Gallego, C.; Johansson, D.P.; Scheers, N.M.; Marklinder, I.M.; Eriksen, A.K.; Silventoinen, P.C.; Nordlund, E.; Sozer, N.; et al. Grains—A Major Source of Sustainable Protein for Health. Nutr. Rev. 2022, 80, 1648–1663. [Google Scholar] [CrossRef]
- Gupta, R.K.; Gangoliya, S.S.; Singh, N.K. Reduction of Phytic Acid and Enhancement of Bioavailable Micronutrients in Food Grains. J. Food Sci. Technol. 2015, 52, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jood, S. Effect of Germination and Probiotic Fermentation on PH, Titratable Acidity, Dietary Fibre, β-Glucan and Vitamin Content of Sorghum Based Food Mixtures. J. Nutr. Food Sci. 2012, 02, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Obafemi, Y.D.; Oranusi, S.U.; Ajanaku, K.O.; Akinduti, P.A.; Leech, J.; Cotter, P.D. African Fermented Foods: Overview, Emerging Benefits, and Novel Approaches to Microbiome Profiling. NPJ Sci. Food 2022, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Zain, M.E. Impact of Mycotoxins on Humans and Animals. J. Saudi Chem. Soc. 2011, 15, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Wafula, E.N.; Muhonja, C.N.; Kuja, J.O.; Owaga, E.E.; Makonde, H.M.; Mathara, J.M.; Kimani, V.W. Lactic Acid Bacteria from African Fermented Cereal-Based Products: Potential Biological Control Agents for Mycotoxins in Kenya. J. Toxicol. 2022, 2022, 2397767. [Google Scholar] [CrossRef] [PubMed]
- Meijer, N.; Kleter, G.; de Nijs, M.; Rau, M.-L.; Derkx, R.; van der Fels-Klerx, H.J. The Aflatoxin Situation in Africa: Systematic Literature Review. Compr. Rev. Food Sci. Food Saf. 2021, 20, 2286–2304. [Google Scholar] [CrossRef]
- Leeuwendaal, N.K.; Stanton, C.; O’Toole, P.W.; Beresford, T.P. Fermented Foods, Health and the Gut Microbiome. Nutrients 2022, 14, 1527. [Google Scholar] [CrossRef]
- Marco, M.L.; Heeney, D.; Binda, S.; Cifelli, C.J.; Cotter, P.D.; Foligné, B.; Gänzle, M.; Kort, R.; Pasin, G.; Pihlanto, A.; et al. Health Benefits of Fermented Foods: Microbiota and Beyond. Curr. Opin. Biotechnol. 2017, 44, 94–102. [Google Scholar] [CrossRef]
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular Analysis of Commensal Host-Microbial Relationships in the Intestine. Science 2001, 291, 881–884. [Google Scholar] [CrossRef] [Green Version]
- Drewes, J.L.; Housseau, F.; Sears, C.L. Sporadic Colorectal Cancer: Microbial Contributors to Disease Prevention, Development and Therapy. Br. J. Cancer 2016, 115, 273–280. [Google Scholar] [CrossRef]
- Gold, A.; Zhu, J. Not Just a Gut Feeling: A Deep Exploration of Functional Bacterial Metabolites That Can Modulate Host Health. Gut Microbes 2022, 14, 2125734. [Google Scholar] [CrossRef] [PubMed]
- Lebeer, S.; Vanderleyden, J.; De Keersmaecker, S.C.J. Genes and Molecules of Lactobacilli Supporting Probiotic Action. Microbiol. Mol. Biol. Rev. 2008, 72, 728–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leech, J.; Cabrera-Rubio, R.; Walsh, A.M.; Macori, G.; Walsh, C.J.; Barton, W.; Finnegan, L.; Crispie, F.; O’Sullivan, O.; Claesson, M.J.; et al. Fermented-Food Metagenomics Reveals Substrate-Associated Differences in Taxonomy and Health-Associated and Antibiotic Resistance Determinants. mSystems 2020, 5, e00522-20. [Google Scholar] [CrossRef] [PubMed]
- Deka, P.; Mehetre, G.T.; Lalnunmawii, E.; Upadhyaya, K.; Singh, G.; Hashem, A.; Al-Arjani, A.-B.F.; Fathi Abd_Allah, E.; Singh, B.P. Metagenomic Analysis of Bacterial Diversity in Traditional Fermented Foods Reveals Food-Specific Dominance of Specific Bacterial Taxa. Fermentation 2021, 7, 167. [Google Scholar] [CrossRef]
- Anyogu, A.; Olukorede, A.; Anumudu, C.; Onyeaka, H.; Areo, E.; Adewale, O.; Odimba, J.N.; Nwaiwu, O. Microorganisms and Food Safety Risks Associated with Indigenous Fermented Foods from Africa. Food Control 2021, 129, 108227. [Google Scholar] [CrossRef]
- Soro-Yao, A.A.; Brou, K.; Amani, G.; Thonart, P.; Djè, K.M. The Use of Lactic Acid Bacteria Starter Cultures during the Processing of Fermented Cereal-Based Foods in West Africa: A Review. Trop. Life Sci. Res. 2014, 25, 81–100. [Google Scholar]
- Chileshe, J.; van den Heuvel, J.; Handema, R.; Zwaan, B.J.; Talsma, E.F.; Schoustra, S. Nutritional Composition and Microbial Communities of Two Non-Alcoholic Traditional Fermented Beverages from Zambia: A Study of Mabisi and Munkoyo. Nutrients 2020, 12, 1628. [Google Scholar] [CrossRef]
- Parker, M.; Zobrist, S.; Donahue, C.; Edick, C.; Mansen, K.; Nadjari, M.H.Z.; Heerikhuisen, M.; Sybesma, W.; Molenaar, D.; Diallo, A.M.; et al. Naturally Fermented Milk From Northern Senegal: Bacterial Community Composition and Probiotic Enrichment With Lactobacillus Rhamnosus. Front. Microbiol. 2018, 9, 2218. [Google Scholar] [CrossRef] [Green Version]
- Ezekiel, C.N.; Ayeni, K.I.; Ezeokoli, O.T.; Sulyok, M.; van Wyk, D.A.B.; Oyedele, O.A.; Akinyemi, O.M.; Chibuzor-Onyema, I.E.; Adeleke, R.A.; Nwangburuka, C.C.; et al. High-Throughput Sequence Analyses of Bacterial Communities and Multi-Mycotoxin Profiling During Processing of Different Formulations of Kunu, a Traditional Fermented Beverage. Front. Microbiol. 2019, 9, 3282. [Google Scholar] [CrossRef]
- Schmidhuber, J.; Tubiello, F.N. Global Food Security under Climate Change. Proc. Natl. Acad. Sci. USA 2007, 104, 19703–19708. [Google Scholar] [CrossRef] [Green Version]
- Diaz, M.; Kellingray, L.; Akinyemi, N.; Adefiranye, O.O.; Olaonipekun, A.B.; Bayili, G.R.; Ibezim, J.; du Plessis, A.S.; Houngbédji, M.; Kamya, D.; et al. Comparison of the Microbial Composition of African Fermented Foods Using Amplicon Sequencing. Sci. Rep. 2019, 9, 13863. [Google Scholar] [CrossRef] [PubMed]
- Chibuzor-Onyema, I.E.; Ezeokoli, O.T.; Sulyok, M.; Notununu, I.; Petchkongkaew, A.; Elliott, C.T.; Adeleke, R.A.; Krska, R.; Ezekiel, C.N. Metataxonomic Analysis of Bacterial Communities and Mycotoxin Reduction during Processing of Three Millet Varieties into Ogi, a Fermented Cereal Beverage. Food Res. Int. 2021, 143, 110241. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- PuTTY: A Free SSH and Telnet Client. Available online: https://www.chiark.greenend.org.uk/~sgtatham/putty (accessed on 16 October 2022).
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D.; et al. XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]
- Brown, S.T.; Buitrago, P.; Hanna, E.; Sanielevici, S.; Scibek, R.; Nystrom, N.A. Bridges-2: A Platform for Rapidly-Evolving and Data Intensive Research. In Proceedings of the Practice and Experience in Advanced Research Computing, Boston, MA, USA, 17 July 2021; pp. 1–4. [Google Scholar]
- Foster, I. Globus Online: Accelerating and Democratizing Science through Cloud-Based Services. IEEE Internet Comput. 2011, 15, 70–73. [Google Scholar] [CrossRef]
- Downloading SRA Toolkit Ncbi/Sra-Tools Wiki. Available online: https://github.com/ncbi/sra-tools (accessed on 16 October 2022).
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 17 October 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Faith, D.P. Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An Improved Greengenes Taxonomy with Explicit Ranks for Ecological and Evolutionary Analyses of Bacteria and Archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) Format or: How I Learned to Stop Worrying and Love the Ome-Ome. GigaScience 2012, 1, 2047-217X. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Pswarayi, F.; Gänzle, M. African Cereal Fermentations: A Review on Fermentation Processes and Microbial Composition of Non-Alcoholic Fermented Cereal Foods and Beverages. Int. J. Food Microbiol. 2022, 378, 109815. [Google Scholar] [CrossRef]
- Zapaśnik, A.; Sokołowska, B.; Bryła, M. Role of Lactic Acid Bacteria in Food Preservation and Safety. Foods 2022, 11, 1283. [Google Scholar] [CrossRef]
- Owade, J.O.; Abong’, G.O.; Okoth, M.W.; Mwang’ombe, A.W.; Jobor, J.O. Comparative Profiling of Lactic Acid Bacteria Isolates in Optimized and Spontaneous Fermentation of Cowpea Leaves. Food Sci. Nutr. 2021, 9, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Durack, J.; Lynch, S.V. The Gut Microbiome: Relationships with Disease and Opportunities for Therapy. J. Exp. Med. 2019, 216, 20–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [Green Version]
- Holzapfel, W.H.; Haberer, P.; Geisen, R.; Björkroth, J.; Schillinger, U. Taxonomy and Important Features of Probiotic Microorganisms in Food and Nutrition. Am. J. Clin. Nutr. 2001, 73, 365S–373S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sade, N.; Peleg, Z. Future Challenges for Global Food Security under Climate Change. Plant Sci. 2020, 295, 110467. [Google Scholar] [CrossRef]
- Wheeler, T.; von Braun, J. Climate Change Impacts on Global Food Security. Science 2013, 341, 508–513. [Google Scholar] [CrossRef]
- Lupton, F.G.H. Lost Crops of Africa: Volume 1. Exp. Agric. 1997, 33, 247–252. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Lactobacillales Genus | Diaz et. al. [31] | Ezekiel et. al. [29] | Chibuzor-Onyema et. al. [32] |
|---|---|---|---|
| Lactobacillus | ***** | ** | ******** |
| Lactococcus | + | + | + |
| Leuconostoc | + | + | |
| Pediococcus | + | + | **** |
| Streptococcus | + | + | |
| Aerococcus | |||
| Alloiococcus | |||
| Carnobacterium | |||
| Dolosigranulum | |||
| Enterococcus | ** | ||
| Oenococcus | |||
| Tetragenococcus | |||
| Vagococcus | |||
| Weissella | + | + | * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dania, M.I.; Faraji, B.; Wachira, J. Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes. Int. J. Environ. Res. Public Health 2022, 19, 16621. https://doi.org/10.3390/ijerph192416621
Dania MI, Faraji B, Wachira J. Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes. International Journal of Environmental Research and Public Health. 2022; 19(24):16621. https://doi.org/10.3390/ijerph192416621
Chicago/Turabian StyleDania, Margaret I., Bahram Faraji, and James Wachira. 2022. "Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes" International Journal of Environmental Research and Public Health 19, no. 24: 16621. https://doi.org/10.3390/ijerph192416621
APA StyleDania, M. I., Faraji, B., & Wachira, J. (2022). Micronutrient Biosynthesis Potential of Spontaneous Grain Fermentation Microbiomes. International Journal of Environmental Research and Public Health, 19(24), 16621. https://doi.org/10.3390/ijerph192416621