
Is a PCSK9 Inhibitor Right for Your Patient? A Review of Treatment Data for Individualized Therapy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Background
2.1. Statin Association with Myopathy
2.2. Statins and Myocardium
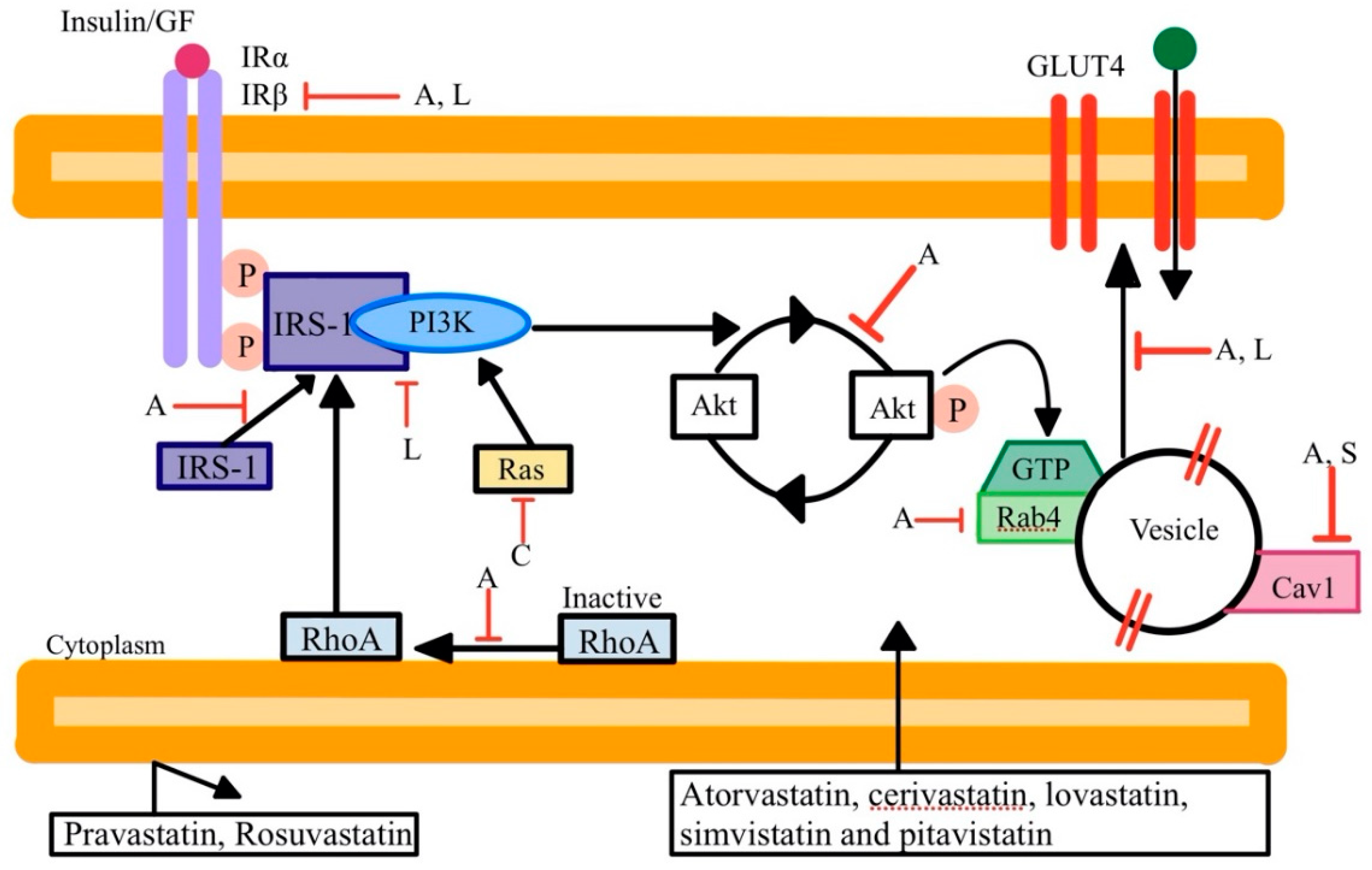
2.3. Statin Association with Diabetes
2.4. Statin-Resistant Hyperlipidemia
3. Review of Non-Statin Treatment Options
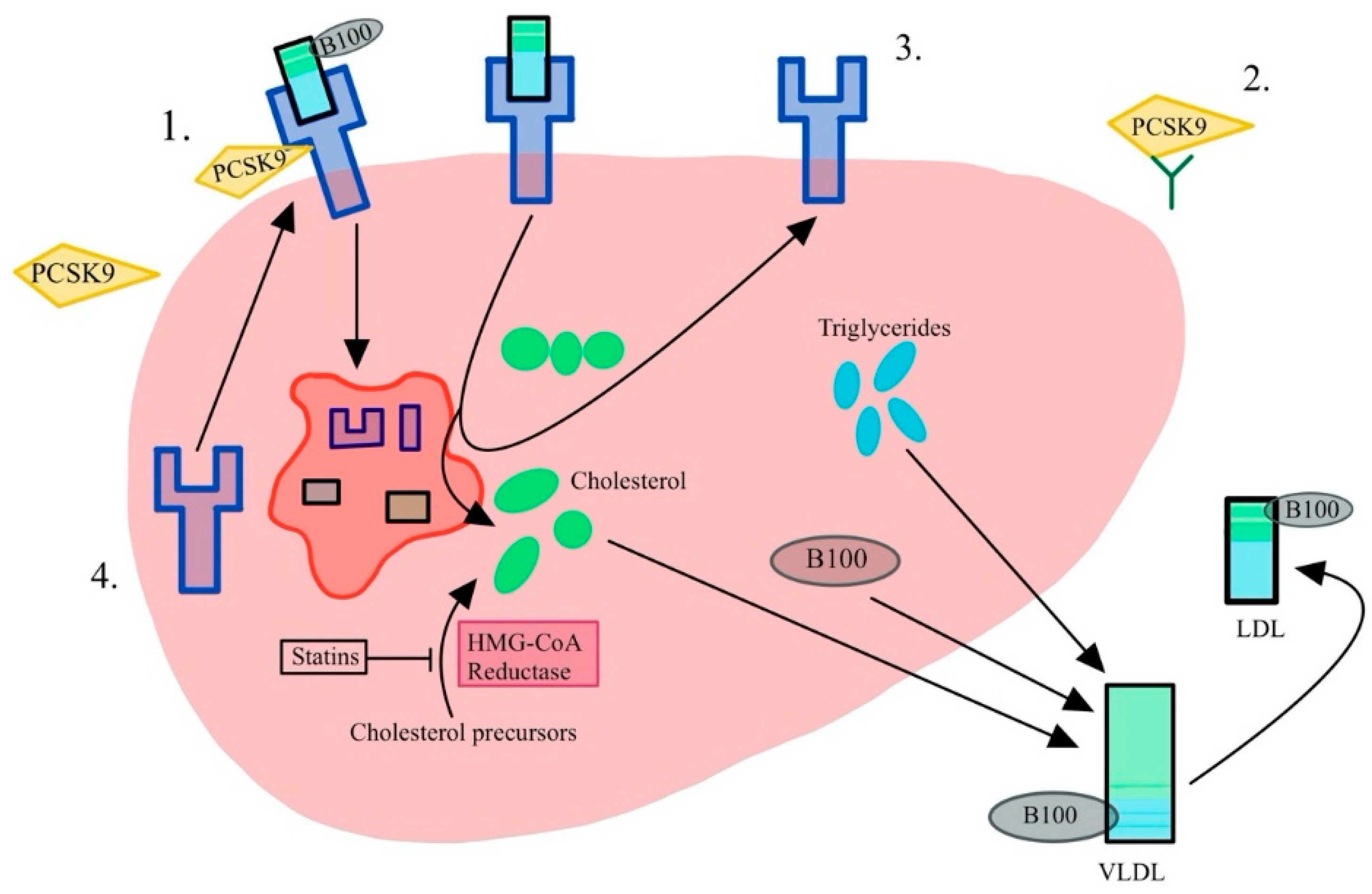
3.1. PCSK9 Inhibitors
3.2. Atheroprotective Properties of PCSK9 Inhibitors
3.3. Ezetimibe
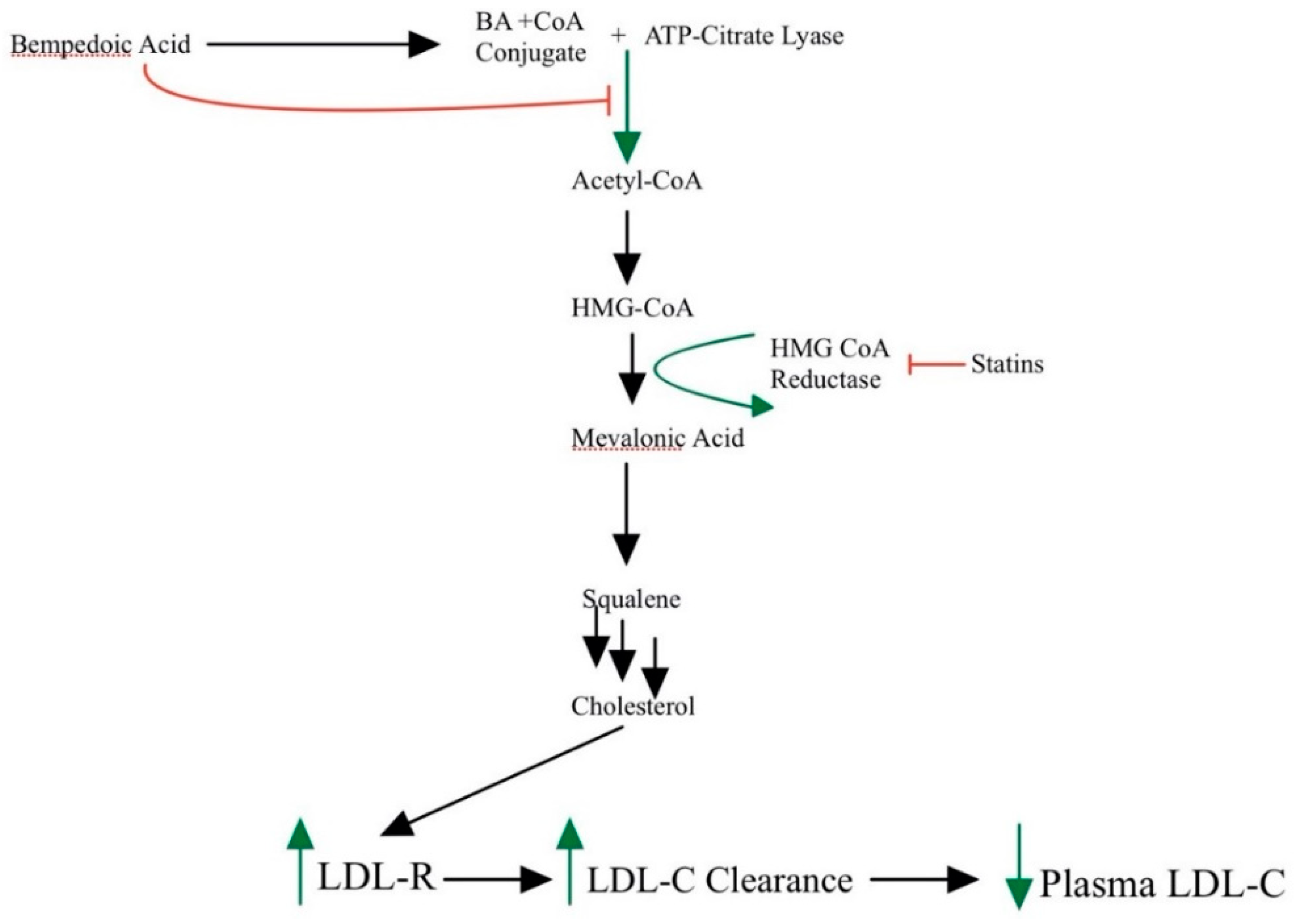
3.4. Bempedoic Acid
3.5. Red Yeast Rice
3.6. Inclisiran
4. Methods
4.1. Systematic Search Strategy
4.2. Systematic Review of 9 Meta-Analyses
4.3. Statistical Analysis
5. Results
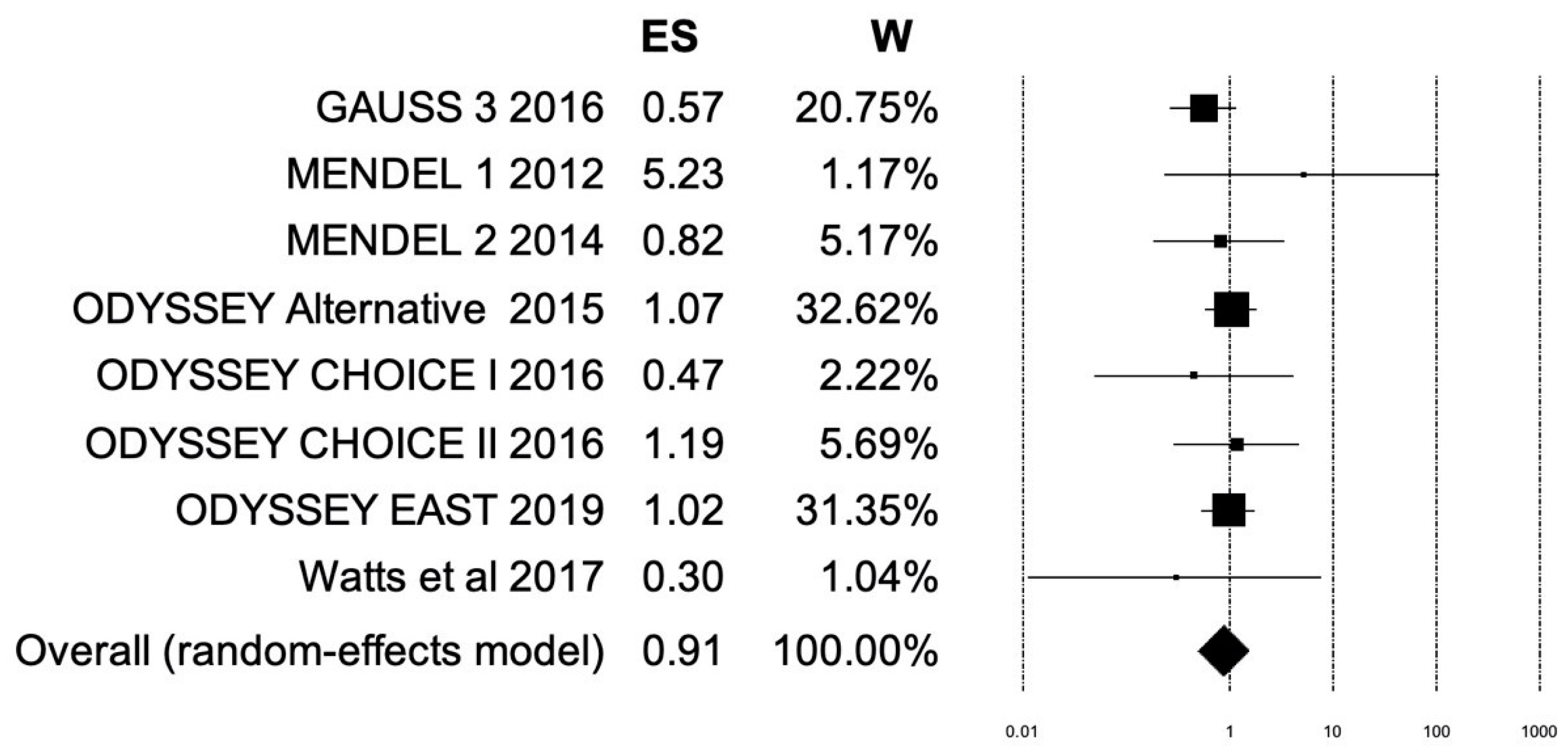
5.1. Results of Meta-Analysis: Myalgia
5.2. Results of Meta-Analysis: Myocardial Infarction
6. Discussion
6.1. Clinical Significance
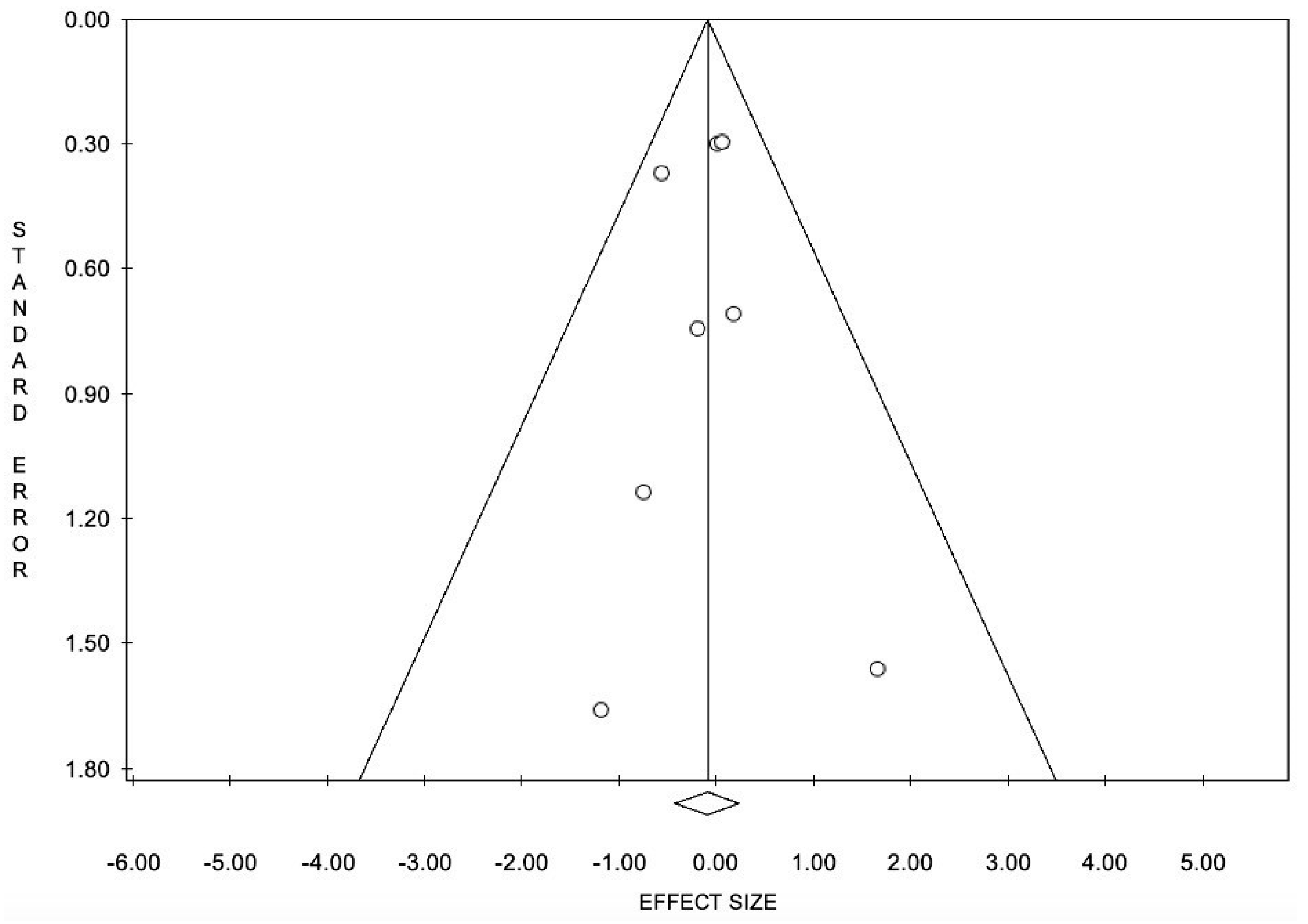
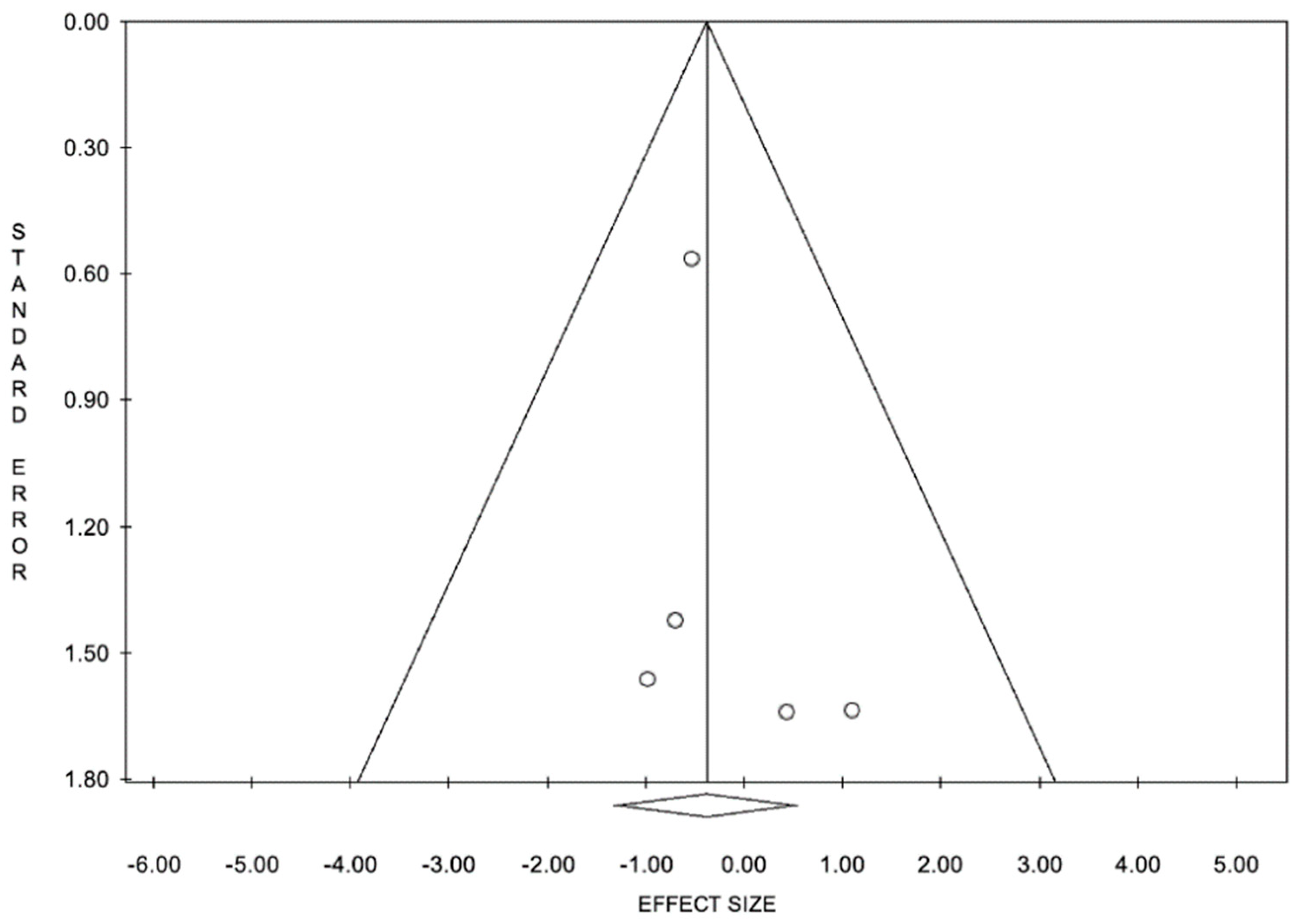
6.2. Limitations and Remarks
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karr, S. Epidemiology and management of hyperlipidemia. Am. J. Manag. Care 2017, 23 (Suppl. S9), S139–S148. [Google Scholar]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A. Guideline on the primary prevention of cardiovascular disease. J. Am. Coll. Cardiol. 2019, 74, e177–e232. [Google Scholar] [CrossRef] [PubMed]
- Branchi, A.; Fiorenza, A.M.; Rovellini, A.; Torri, A.; Muzio, F.; Macor, S.; Sommariva, D. Lowering effects of four different statins on serum triglyceride level. Eur. J. Clin. Pharm. 1999, 55, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fu, J.; Koonen, D.; Kuivenhoven, J.A.; Snieder, H.; Hofker, M.H. Are hypertriglyceridemia and HDL causal factors in the development of insulin resistance? Atherosclerosis 2014, 230, 130–138. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Kostantinou, A.; Kougias, M.; Kazazis, C. Statins and cancer. Anticancer Agents Med. Chem. 2014, 14, 706–712. [Google Scholar] [CrossRef]
- Chang, H.L.; Chen, C.Y.; Hsu, Y.F.; Kuo, W.S.; Ou, G.; Chiu, P.T.; Huang, Y.H.; Hsu, M.J. Simvastatin induced HCT116 colorectal cancer cell apoptosis through p38MAPK-p53-survivin signaling cascade. Biochim. Biophys. Acta 2013, 8, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Dubuc, G.; Chamberland, A.; Wassef, H.; Davignon, D.; Seidah, N.G.; Bernier, L.; Prat, A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2004, 8, 1454–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.M.Y.; Sattar, N.; McMurray, J.J.V.; Packard, C.J. Statins in the Prevention and Treatment of Heart Failure: A Review of the Evidence. Curr. Atheroscler. Rep. 2019, 21, 41. [Google Scholar] [CrossRef] [Green Version]
- Charach, G.; Argov, O.; Nochomovitz, H.; Rogowski, O.; Charach, L.; Grosskopf, I. A longitudinal 20 years of follow up showed a decrease in the survival of heart failure patients who maintained low LDL cholesterol levels. QJM 2018, 11, 319–325. [Google Scholar] [CrossRef]
- Yoon, D.; Sheen, S.S.; Lee, S.; Choi, Y.J.; Park, R.W.; Lim, H.S. Statins and risk for new-onset diabetes mellitus: A real-world cohort study using a clinical research database. Medicine 2016, 95, e5429. [Google Scholar] [CrossRef]
- Choi, J.Y.; Choi, C.U.; Hwang, S.Y.; Choi, B.G.; Jang, W.Y.; Kim, D.Y. Effect of pitavastatin compared with atorvastatin and rosuvastatin on new-onset diabetes mellitus in patients with acute myocardial infarction. Am. J. Cardiol. 2018, 122, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Cederberg, H.; Stančáková, A.; Yaluri, N.; Modi, S.; Kuusisto, J.; Laakso, M. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: A 6 year follow-up study of the METSIM cohort. Diabetologia 2015, 58, 1109–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiman, U.; Najmi, A.; Khan, R. Statin induced diabetes and its clinical implications. J. Pharmacol. Pharmacother. 2014, 5, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J. Statins and diabetes risk: How real is it and what are the mechanisms? Curr. Opin. Lipidol. 2015, 26, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Cho, L.; Dent, R.; Stroes, E.S.G.; Stein, E.A.; Sullivan, D.; Ruzza, A.; Flower, A.; Somaratne, R.; Rosenson, R.S. Persistent safety and efficacy of evolocumab in patients with statin intolerance: A subset analysis of the OSLER open-label extension studies. Cardiovasc. Drugs Ther. 2018, 32, 365–372. [Google Scholar] [CrossRef]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Wilke, R.A.; Ramsey, L.B.; Johnson, S.B.; Maxwell, W.D.; McLead, H.L.; Voora, D.; Krauss, R.M.; Roden, D.M.; Feng, Q.; Cooper-Dehoff, R.M.; et al. The clinical pharmacogenomics implementation consortium. CPIC guideline for SLCO1B1 and simavastatin-induced myopathy. Clin. Pharmacol. Ther. 2012, 92, 112–117. [Google Scholar] [CrossRef]
- Kee, P.S.; Chin, P.K.L.; Kennedy, M.A.; Maggo, S.D.S. Pharmacogenetics of statin-induced myotoxicity. Front. Genet. 2020, 11, 575678. [Google Scholar] [CrossRef]
- Isackson, P.J.; Ochs-Balcom, H.M.; Ma, C.; Harley, J.B.; Peltier, W.; Tarnopolsky, M.; Sripathi, M.; Wortmann, R.L.; Simmons, Z.; Wilson, J.D. Association of common variants in the human eyes shut ortholog (EYS) with statin-induced myopathy: Evidence for additionl functions of EYS. Muscle Nerve 2011, 44, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Vrablik, M.; Hubácek, J.A.; Dlouha, D.; Lanska, V.; Rynekrova, J.; Zlatohlavek, L.; Prusikova, M.; Ceska, R.; Adamkova, V. Impact of variants within seven candidate genes on statin treatment efficacy. Physiol. Res. 2012, 61, 609–617. [Google Scholar] [CrossRef]
- Derosa, G.; D’Angelo, A.; Maffioli, P. Coenzyme q10 liquid supplementation in dyslipidemic subjects with statin-related clinical symptoms: A double-blind, randomized, placebo-controlled study. Drug Des. Devel. Ther. 2019, 13, 3647–3655. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Guo, M.; Chai, H.; Wang, W.T.; Gao, Z.Y.; Shi, D.Z. Effects of coenzyme Q10 on statin-induced myopathy: An updated meta-analysis of randomized controlled trials. J. Am. Heart Assoc. 2018, 7, e009835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierno, S.; Camerino, G.M.; Cippone, V.; Rolland, J.-F.; Desaphy, J.-F.; De Luca, A.; Liantonio, A.; Bianco, G.; Kunic, J.D.; George, A.L., Jr.; et al. Statins and fenofibrate affect skeletal muscle chloride conductance in rats by differently impairing ClC-1 channel regulation and expression. Br. J. Pharmacol. 2009, 156, 1206–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuy, M.C.; Preziosi, P.; Maamer, M.; Arnaud, S.; Galan, P.; Hercberg, S.; Meunier, P.J. Prevalence of vitamin D insufficiency in an adult normal population. Osteoporos. Int. 1997, 7, 439–443. [Google Scholar] [CrossRef]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Hassan Murad, M.; Weaver, C.M.; Endocrine Society. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, W.; Khan, N.; Glueck, C.J.; Pandey, S.; Wang, P.; Goldenberg, N.; Uppal, M.; Khanal, S. Low serum 25 (OH) vitamin D levels (<32 ng/mL) are associated with reversible myositis-myalgia in statin-treated patients. Transl. Res. 2009, 153, 11–16. [Google Scholar] [CrossRef]
- Bell, D.S. Resolution of statin-induced myalgias by correcting vitamin D deficiency. S. Med. J. 2010, 103, 690–692. [Google Scholar] [CrossRef]
- Lee, P.; Greenfield, J.R.; Campbell, L.V. Vitamin D insufficiency–a novel mechanism of statin-induced myalgia? Clin. Endocrinol. 2009, 71, 154–155. [Google Scholar] [CrossRef]
- Khayznikov, M.; Hemachrandra, K.; Pandit, R.; Kumar, A.; Wang, P.; Glueck, C.J. Statin Intolerance Because of Myalgia, Myositis, Myopathy, or Myonecrosis Can in Most Cases be Safely Resolved by Vitamin D Supplementation. N. Am. J. Med. Sci. 2015, 7, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Riche, K.D.; Arnall, J.; Rieser, K.; East, H.E.; Riche, D.M. Impact of vitamin D status on statin-induced myopathy. J. Clin. Transl. Endocrinol. 2016, 6, 56–59. [Google Scholar] [CrossRef]
- King, M.; Kingery, J.; Casey, B. Diagnosis and evaluation of heart failure. Am. Fam. Physician 2012, 85, 1161–1168. [Google Scholar] [PubMed]
- Bozkurt, B.; Coats, A.J.S.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Bohm, M.; Butler, J.; et al. Universal Definition and Classification of Heart Failure. A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing committee of the Universal Definition of Heart Failure. J. Card. Fail. 2021, 27, 387–413. [Google Scholar] [CrossRef]
- Sola, S.; Mir, M.; Lerakis, S.; Tandon, N.; Khan, B.V. Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure. J. Am. Coll. Card. 2006, 47, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Koh, K.K. Effects of statins on vascular wall: Vasomotor function, inflammation and plaque stability. Cardiovasc. Res. 2000, 47, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Ford, I.; Shah, A.S.; Zhang, R.; McAllister, D.A.; Strachan, F.A.; Gaslake, M.; Newby, D.E.; Packard, C.J.; Mill, N.L. High-Sensitivity Cardiac Troponin, Statin Therapy, and Risk of Coronary Heart Disease. J. Am. Coll. Cardiol. 2016, 68, 2719–2728. [Google Scholar] [CrossRef] [PubMed]
- Galacia-Garcia, U.; Jebari, S.; Larrea-Sebal, A.; Uribe, K.B.; Siddiqi, H.; Ostolaza, H.; Benito-Vicente, A.; Martin, C. Statin treatment-induced development of type 2 diabetes: From clinical evidence to mechanistic insights. Int. J. Mol. Sci. 2020, 21, 4725. [Google Scholar] [CrossRef] [PubMed]
- Kjekshus, J.; Apetrei, E.; Barrios, V.; Bohm, M.; Cleland, G.F.; Cornel, J.H.; Dunselman, P.; Fonseca, A.; Goudev, A.; Grande, P.; et al. Rosuvastatin in Older Patients with Systolic Heart Failure. N. Engl. J. Med. 2007, 357, 2248–2261. [Google Scholar] [CrossRef]
- Rallidis, L.; Skoumas, I.; Liberopoulos, E.; Vlachopoulos, C.; Kiourri, E.; Koutagiar, I.; Anastasiou, G.; Kosmas, N.; Elisaf, M.S.; Tousoulis, D.; et al. PCSK9 inhibitors in clinical practice: Novel directions and new experiences. Hellenic. J. Cardiol. 2020, 61, 241–245. [Google Scholar] [CrossRef]
- Koren, M.J.; Scott, R.; Kim, J.B.; Knusel, B.; Liu, T.; Lei, L.; Bolognese, M.; Wasserman, S.M. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): A randomized, double-blind, placebo-controlled phase 2 study. Lancet 2012, 380, 1995–2006. [Google Scholar] [CrossRef]
- Koren, M.J.; Lundqvist, P.; Bolognese, M.; Neutel, J.M.; Monsalvo, M.L.; Yang, J.; Kim, J.B.; Scott, R.; Wasserman, S.W.; Bays, H.; et al. Anti-pcsk9 monotherapy for hypercholesterolemia. The MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2531–2540. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.F.; Hyde, C.L.; Wood, L.S.; Paciga, S.A.; Hinds, D.A.; Cox, D.R.; Kees Hovingh, G.; Kastelein, J.J.P. Comprehensive whole-genome and candidate gene analysis for response to statin therapy in the Treating to New Targets (TNT) cohort. Circ. Cardiovasc. Genet. 2009, 2, 173–181. [Google Scholar] [CrossRef]
- Peters, B.J.M.; Pett, H.; Klungel, O.H.; Stricker, B.H.C.; Psaty, B.M.; Glazer, N.L.; Wiggins, K.L.; Bis, J.C.; de Boer, A. Genetic variability within the cholesterol lowering pathway and the effectiveness of statins in reducing the risk of MI. Atherosclerosis 2011, 217, 458–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.N.; Ballantyne, C.M.; Gotto Jr, A.M.; Tan, Y.; Willerson, J.T.; Marian, A.J. A common PCSK9 haplotype, encompassing the E670G coding single nucleotide polymorphism, is a novel genetic marker for plasma low-density lipoprotein cholesterol levels and severity of coronary atherosclerosis. J. Am. Coll. Cardiol. 2005, 45, 1611–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abboud, S.; Karhunen, P.J.; Lutjohann, D.; Goebeler, S.; Luoyo, T.; Friedrichs, S.; Lehtimaki, T.; Pandolfo, M.; Laaksonen, R. Proprotein convertase subtililsin/kenix type 9 (PCSK9) gene is a risk factor of large-vessel atherosclerosis stroke. PLoS ONE 2007, 2, e1043. [Google Scholar] [CrossRef] [PubMed]
- Kotlega, D.; Golab-Janowska, M.; Masztalewicz, M.; Ciecwiez, S.; Nowacki, P. Association between selected gene polymorphisms and statin metabolism, risk of ischemic stroke and cardiovascular disorders. Postep. Hig. Med. Dosw. 2016, 70, 435–447. [Google Scholar] [CrossRef]
- Anderson, J.M.; Cerda, A.; Hirata, M.H.; Rodrigues, A.C.; Dorea, E.L.; Bernik, M.M.S.; Bertolami, M.C.; Faludi, A.A.; Hirata, R.D.C. Influence of PCSK9 polymorphisms on plasma lipids and response to atorvastatin treatment in Brazilian subjects. J. Clin. Lipidol. 2014, 8, 256–264. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Awan, Z.; Seidah, N.G.; MacFadyen, J.G.; Benjannet, S.; Chasman, D.I.; Ridker, P.M.; Genest, J. Rosuvastatin, proprotein convertase subtilisin/Kexin type 9 concentrations, and LDL cholesterol response: The JUPITER trial. Clin. Chem. 2012, 58, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Watts, G.F.; Chan, D.C.; Dent, R.; Somaratne, R.; Wasserman, S.M.; Scott, R.; Burrows, S.; Barrett, P.H.R. Factorial effects of evolocumab and atorvastatin on lipoprotein on metabolism. Circulation 2017, 135, 338–351. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Turgeon, R.; Tsuyuki, R.; Gyenes, G.; Pearson, G. Cardiovascular efficacy and safety of PCSK9 inhibitors: Systematic review and meta-analysis including the ODYSSEY OUTCOMES trial. Can. J. Cardiol. 2018, 34, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, J.; Chopra, V.K.; Zhang, S.; Su, G.; Ma, C.; Huang, Z.; Ma, Y.; Yao, Z.; Yuan, Z.; et al. ODDYSEY EAST: Alirocumab efficacy and safety vs ezetimibe in high cardiovascular risk patients with hypercholesterolemia and on maximally tolerated statin in China, India, and Thailand. J. Clin. Lipidol. 2020, 14, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarty, P.M.; Thompson, P.D.; Cannon, C.P.; Guyton, J.R.; Bergeron, J.; Zieve, F.J.; Bruckert, E.; Jacobson, T.A.; Kopecky, S.L.; Baccara-Dinet, M.T.; et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The ODYSSEY ALTERNATIVE randomized trial. J. Clin. Lipidol. 2015, 9, 758–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, D.; Olsson, A.G.; Scott, R.; Kim, J.B.; Xue, A.; Gebski, V.; Wasserman, S.M.; Stein, E.A. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: The GAUSS randomized trial. JAMA 2012, 308, 2497–2506. [Google Scholar] [CrossRef] [Green Version]
- Stroes, E.; Colquhoun, D.; Sullivan, D.; Civeira, F.; Rosenson, R.S.; Watts, G.F.; Bruckert, E.; Cho, L.; Dent, R.; Knusel, B.; et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: The GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2541–2548. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E.; Stroes, E.; Dent-Acosta, R.E.; Rosenson, R.S.; Lehman, S.J.; Sattar, N.; Preiss, D.; Bruckert, E.; Ceska, R.; Lepor, L.; et al. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle-related statin intolerance: The GAUSS-3 randomized clinical trial. JAMA 2016, 315, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Koba, S.; Inoue, I.; Cyrille, M.; Lu, C.; Inomata, H.; Smimauchi, J.; Kajinami, K. Evolocumab vs. ezetimibe in statin-intolerant hyperlipidemic Japanese patients: Phase 3 GAUSS-4 trial. J. Atheroscler. Thromb. 2020, 27, 471–484. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, M.L.; Giugliano, R.P.; Wiviott, S.D.; Atar, D.; Keech, A.; Kuder, J.F.; Im, K.; Murphy, S.A.; Flores-Arredondo, J.H.; Lopez, J.A.G.; et al. Long-term evolocumab in patients with established atherosclerotic cardiovascular disease. Circulation 2022, 146, 1109–1119. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Ceska, R.; et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef]
- Casula, M.; Olmastroni, E.; Boccalari, M.T.; Tragni, E.; Pirillo, A.; Catapano, A.L. Cardiovascular events with PCSK9 inhibitors: An updated meta-analysis of randomized controlled trials. Pharmacol. Res. 2019, 143, 143–150. [Google Scholar] [CrossRef]
- Monami, M.; Sesti, G.; Mannucci, E. PCSK9 inhibitor therapy: A systematic review and meta-analysis of metabolic and cardiovascular outcomes in patients with diabetes. Diabetes Obes. Metab. 2019, 21, 903–908. [Google Scholar] [CrossRef]
- Dicembrini, I.; Giannini, S.; Ragghianti, B.; Mannucci, E.; Monami, M. Effects of PCSK9 inhibitors on LDL cholesterol, cardiovascular morbidity and all-cause mortality: A systematic review and meta-analysis of randomized controlled trials. J. Endocrinol. Investig. 2019, 42, 1029–1039. [Google Scholar] [CrossRef]
- Karatasakis, A.; Danek, B.; Karacsonyi, J.; Rangan, B.V.; Roesle, M.K.; Knickelbine, T.; Miedema, M.D.; Khalili, H.; Ahmad, Z.; Abdullah, S.; et al. Effect of PCSK9 inhibitors on clinical outcomes in patients with hypercholesterolemia: A meta-analysis of 35 randomized controlled trials. J. Am. Heart Assoc. 2017, 6, e006910. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, L.S.F.; Campos, A.M.; Sposito, A.C. Propotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and incident type 2 diabetes: A systematic review and meta-analysis with over 96,000 patient-years. Diabetes Care 2018, 41, 364–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhu, Q.; Zhu, L.; Chen, J.-Z.; Chen, Q.-H.; Li, G.-N.; Xie, J.; Kang, L.-N.; Xu, B. Safety and efficacy of anti-PCSK9 antibodies: A meta-analysis of 25 randomized, controlled trials. BMC Med. 2015, 13, 123. [Google Scholar] [CrossRef] [PubMed]
- Gouni-Berthold, I.; Descamps, O.S.; Fraass, U.; Hartfield, E.; Allcott, K.; Dent, R.; Marz, W. Systematic review of published phase 3 data on anti-PCSK9 monoclonal antibodies in patients with hypercholesterolemia. Br. J. Clin. Pharmacol. 2016, 82, 1412–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlTurki, A.; Marafi, M.; Dawas, A.; Dube, M.-P.; Vieira, L.; Sherman, M.H.; Gregoire, J.; Thanassoulis, G.; Tardif, J.-C.; Huynh, T. Meta-analysis of randomized controlled trials assessing the impact of proprotein convertase subtilisin/kexin type 9 antibodies on mortality and cardiovascular outcomes. Am. J. Cardiol. 2019, 124, 1869–1875. [Google Scholar] [CrossRef]
- Masson, W.; Lobo, M.; Siniawski, D.; Molinaro, G.; Masson, G.; Huerin, M.; Nogueira, J.P. Role of non-statin lipid-lowering therapy in coronary atherosclerosis regression: A meta-analysis and meta-regression. Lipids Health Dis. 2020, 19, 111. [Google Scholar] [CrossRef]
- Ota, H.; Omori, H.; Kawasaki, M.; Hirakawa, A.; Matsuo, H. Clinical impact of PCSK9 inhibitor on stabilization and regression of lipid-rich coronary plaques: A near-infrared spectroscopy study. Eur. Heart J. Cardiovasc. Imaging 2022, 23, 118–217. [Google Scholar] [CrossRef]
- Raber, L.; Ueki, Y.; Otsuka, T.; Losdat, S.; Haner, J.D.; Lonberg, J.; Fahrni, G.; Iglesias, J.F.; van Geuns, R.-J.; Ondracek, A.S.; et al. Effect of alirocumab added to high-intensity statin therapy on coronary atherosclerosis in patients with acute myocardial infarction: The PACMAN-AMI randomized clinical trial. JAMA 2022, 327, 1771–1781. [Google Scholar] [CrossRef]
- Pouwer, M.; Pieterman, E.; Worms, N.; Keijzer, N.; Jukema, J.W.; Gromada, J.; Gusarova, V.; Princen, H.M.G. Alirocumab, evinacumab, and atorvastatin triple therapy regresses plaque lesions and improves lesion composition in mice. J. Lipid Res. 2020, 61, 365–375. [Google Scholar] [CrossRef]
- Nicholls, S.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.P.; Koenig, W.; Somaratne, R.; Kassahun, H.; Yang, J.; et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: The GLAGOV randomized clinical trial. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Liu, J.; Wang, W.; Wang, M.; Qi, Y.; Zhao, F.; Sun, J.; Liu, J.; Li, Y.; Zhao, D. Association between plasma PCSK9 levels and 10-year progression of carotid atherosclerosis beyond LDL-C: A cohort study. Int. J. Cardiol. 2016, 215, 293–298. [Google Scholar] [CrossRef] [PubMed]
- White, H.D.; Schwartz, G.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Chiang, C.-E.; Diaz, R.; Goodman, S.G.; Jukema, J.W.; Loy, M.; et al. Alirocumab after acute coronary syndrome in patients with a history of heart failure. Eur. Heart J. 2022, 43, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S. PCSK9 inhibitors: Clinical evidence and implementation. Nat. Rev. Cardiol. 2019, 16, 155–165. [Google Scholar] [CrossRef]
- Aranzula, T.; Piazza, S.; Ricotti, A.; Musumeci, G.; Gaggiano, A. CARotid plaqUe StabilizatiOn and regression with evolocumab: Rationale and design of the CARUSO study. Catheter. Cardiovasc. Interv. 2021, 98, E115–E121. [Google Scholar] [CrossRef]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jujema, J.W.; et al. Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef] [Green Version]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M.; CLEAR Harmony Trial. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Goldberg, A.C.; Leiter, L.A.; Stroes, E.S.G.; Baum, S.J.; Hanselman, J.C.; Bloedon, L.T.; Lalwani, N.D.; Patel, P.M.; Zhao, X.; Barton Duell, P. CLEAR Wisdom: Effect of bempedoic acid vs placebo added to maximally tolerated statins on low-density lipoprotein cholesterol in patients at high risk for cardiovascular disease. JAMA 2019, 322, 1780–1788. [Google Scholar] [CrossRef]
- Thompson, P.D.; MacDougall, D.E.; Newton, R.S.; Margulies, J.R.; Hanselman, J.C.; Orloff, D.G.; McKenney, J.M.; Ballantyne, C.M. Treatment with ETC-1002 alone and in combination with ezetimibe lowers LDL cholesterol in hypercholesterolemic patients with or without statin intolerance. J. Clin. Lipidol. 2016, 10, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Ballantyne, C.M.; Banach, M.; Mancini, G.B.J.; Lepor, N.E.; Hanselman, J.C.; Zhao, X.; Leiter, L.A. Efficacy and safety of bempedoic acid added to ezetimibe in statin-intolerant patients with hypercholesterolemia: A randomized placebo-controlled study. Atherosclerosis 2018, 277, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Iskandar, I.; Harahap, Y.; Wijayanti, T.R.; Sandra, M.; Prasaja, B.; Cahyaningsih, P. Efficacy and tolerability of a nutraceutical combination of red yeast rice, guggulipid, and chromium picolinate evaluated in a randomized, placebo- controlled, double-blind study. Complement. Ther. Med. 2020, 48, 102282. [Google Scholar] [CrossRef] [PubMed]
- Fogacci, F.; Banach, M.; Mikhailidis, D.P.; Bruckert, E.; Toth, P.P.; Watts, G.F.; Reiner, Z.; Mancini, J.; Rizzo, M.; Mitchenko, O.; et al. Safety of red yeast rice supplementation: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2019, 143, 1–16. [Google Scholar] [CrossRef]
- USFDA. FDA Approves Add-on Therapy to Lower Cholesterol among Certain High-Risk Adults. 2021. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-add-therapy-lower-cholesterol-among-certain-high-risk-adults (accessed on 22 December 2021).
- Ray, K.K.; Wright, R.S.; Kalland, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two phase 3 trials of inclisiran in patients with elevated ldl cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.; Rossi, J.; Han, S. The current state and future directions of RNA-i based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Azari, S.; Rezapour, A.; Omidi, N.; Alipour, V.; Behzadifar, M.; Safari, H.; Tajdini, M.; Bragazzi, N.L. Cost-effectiveness analysis of PCSK9 inhibitors in cardiovascular diseases: A systematic review. Heart Fail. Rev. 2019, 25, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, E.A.; Main, C.A.; Miracolo, A.; Papadakis, K.; Paparouni, K.; Kanavos, P.G. Potential barriers in lipid-lowering treatment with PCSK9 inhibitors from a healthcare perspective. Comparative evidence from ten countries. Eur. Heart. J. 2022, 43, 2348. [Google Scholar] [CrossRef]
- Cohen, J.D.; Cziraky, M.J.; Jacobson, T.A.; Maki, K.C.; Karalis, D.G. Barriers to PCSK9 inhibitor prescriptions for patients with high cardiovascular risk: Results of a healthcare provider survey conducted by the National Lipid Association. J. Clin. Lipidol. 2017, 11, 891–900. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beltran, R.A.; Zemeir, K.J.; Kimberling, C.R.; Kneer, M.S.; Mifflin, M.D.; Broderick, T.L. Is a PCSK9 Inhibitor Right for Your Patient? A Review of Treatment Data for Individualized Therapy. Int. J. Environ. Res. Public Health 2022, 19, 16899. https://doi.org/10.3390/ijerph192416899
Beltran RA, Zemeir KJ, Kimberling CR, Kneer MS, Mifflin MD, Broderick TL. Is a PCSK9 Inhibitor Right for Your Patient? A Review of Treatment Data for Individualized Therapy. International Journal of Environmental Research and Public Health. 2022; 19(24):16899. https://doi.org/10.3390/ijerph192416899
Chicago/Turabian StyleBeltran, Roman A., Kyle J. Zemeir, Chase R. Kimberling, Mary S. Kneer, Michelle D. Mifflin, and Tom L. Broderick. 2022. "Is a PCSK9 Inhibitor Right for Your Patient? A Review of Treatment Data for Individualized Therapy" International Journal of Environmental Research and Public Health 19, no. 24: 16899. https://doi.org/10.3390/ijerph192416899