
Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Pathogenesis of PCOS
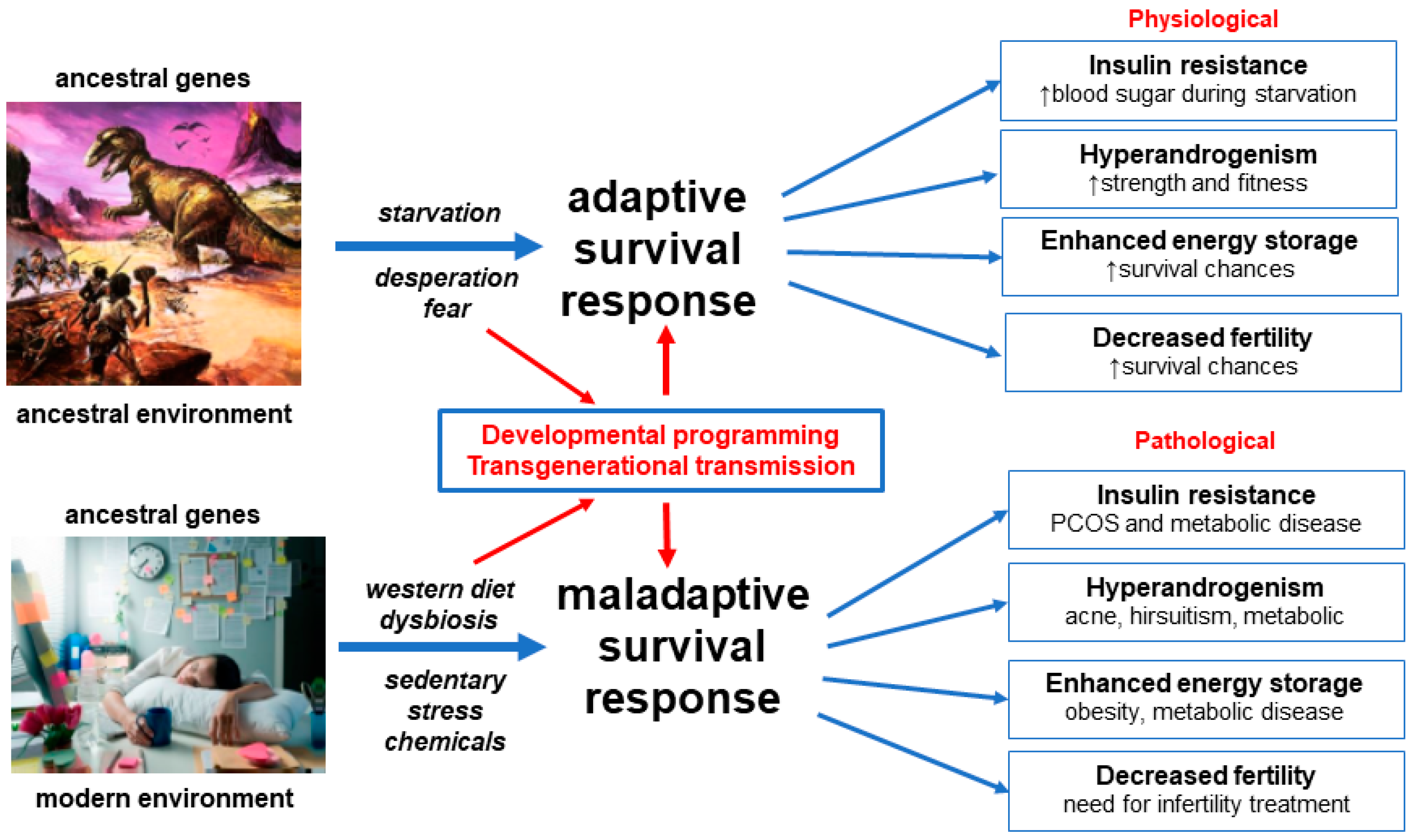
3.1. Evolution
3.2. Genetics
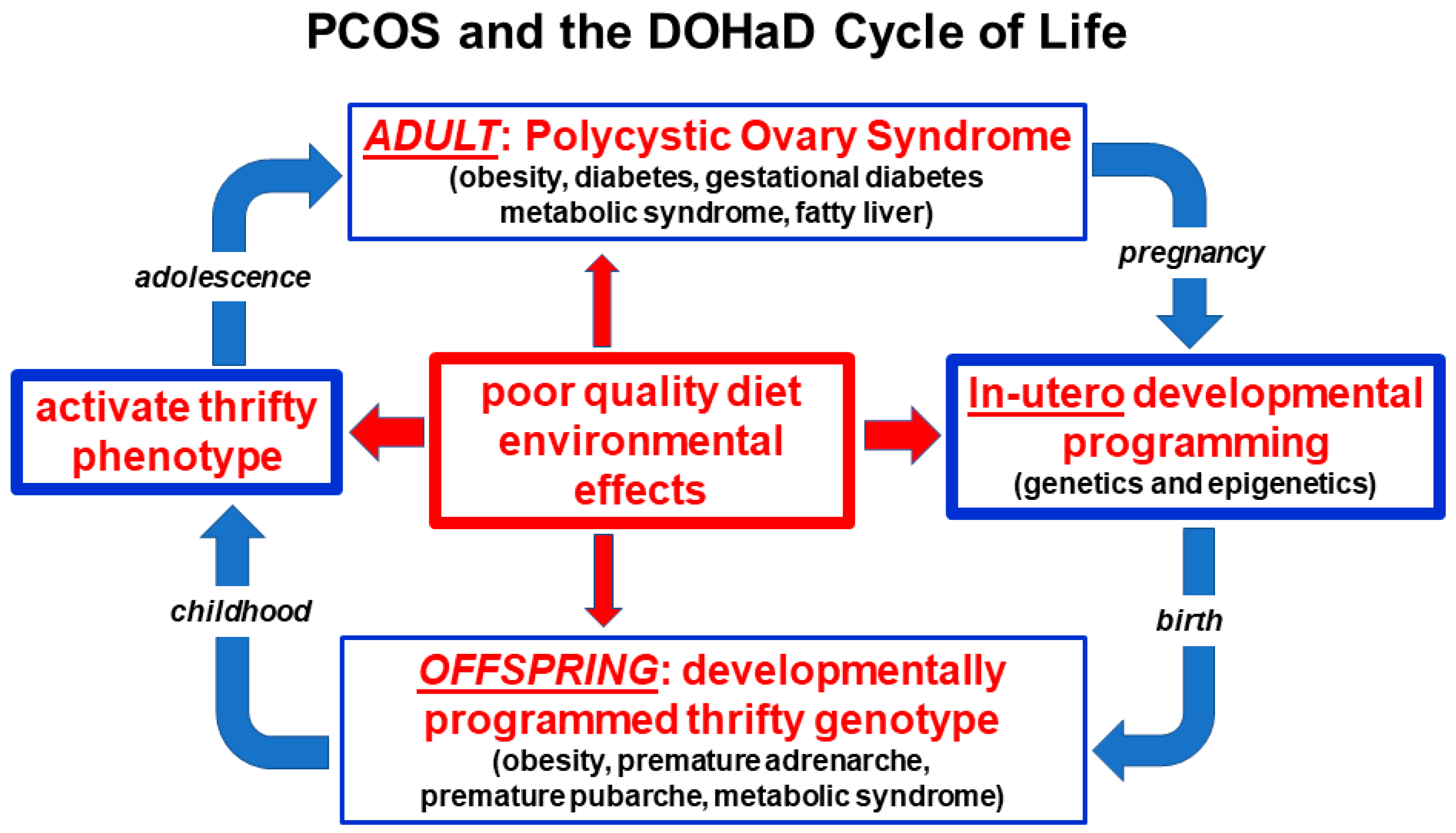
3.3. Developmental Epigenetic Programming
3.4. Microbiome and Dysbiosis
3.5. Insulin Resistance
3.6. Obesity and the Lean PCOS Paradox
3.7. Endocrine-Disrupting Chemical Exposure
3.8. Lifestyle Contributors to the Pathogenesis of PCOS
3.9. Circadian Rhythm Disruption and PCOS
3.10. Conceptual Framework and Summary of the Unified Evolutionary Model
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bodai, B.I.; Nakata, T.E.; Wong, W.T.; Clark, D.R.; Lawenda, S.; Tsou, C.; Liu, R.; Shiue, L.; Cooper, N.; Rehbein, M.; et al. Lifestyle Medicine: A Brief Review of Its Dramatic Impact on Health and Survival. Perm. J. 2017, 22, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMacken, M.; Shah, S. A plant-based diet for the prevention and treatment of type 2 diabetes. J. Geriatr. Cardiol. 2017, 14, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Gakidou, E.; Afshin, A.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulle, A.M.; Abera, S.F.; Aboyans, V.; et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1345–1422. [Google Scholar] [CrossRef] [Green Version]
- Parker, J. NEM: A New Paradigm for Understanding the Common Origins of the Chronic Disease Epidemic. ACNEM J. 2018, 37, 6–11. [Google Scholar]
- Glastras, S.J.; Valvi, D.; Bansal, A. Editorial: Developmental programming of metabolic diseases. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Zore, T.; Joshi, N.V.; Lizneva, D.; Azziz, R. Polycystic Ovarian Syndrome: Long-Term Health Consequences. Semin. Reprod. Med. 2017, 35, 271–281. [Google Scholar] [CrossRef]
- Teede, H.; Misso, M.; Costello, M.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R. International Evidence-Based Guideline for the Assessment and Management of Polycystic Ovary Syndrome 2018; National Health and Medical Research Council [NHMRC]: Canberra, Australia, 2018; pp. 1–198. ISBN 9780646554709.
- Benton, M.L. The influence of evolutionary history on human health and disease. Nat. Rev. Genet. 2021, 22, 269–283. [Google Scholar] [CrossRef]
- Painter, D. The evolution of evolutionary medicine. In The Dynamics of Science: Computational Frontiers in History and Philosophy of Science; Pittsburgh University Press: Pittsburgh, PA, USA, 2020. [Google Scholar]
- Fay, J.C. Disease consequences of human adaptation. Appl. Transl. Genom. 2013, 2, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Pathak, G.; Nichter, M. Polycystic ovary syndrome in globalizing India: An ecosocial perspective on an emerging lifestyle disease. Soc. Sci. Med. 2015, 146, 21–28. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C. Evolutionary and genetic antecedents to the pathogenesis of polycystic ovary syndrome [PCOS]. J. ACNEM 2021, 40, 12–20. [Google Scholar]
- Stearns, S.C. Evolutionary medicine: Its scope, interest and potential. Proc. R. Soc. B Biol. Sci. 2012, 279, 4305–4321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsatsoulis, A.; Mantzaris, M.D.; Sofia, B.; Andrikoula, M. Insulin resistance: An adaptive mechanism becomes maladaptive in the current environment—An evolutionary perspective. Metabolism 2013, 62, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M.A.; Elton, S. Polycystic ovary syndrome: A transgenerational evolutionary adaptation. BJOG Int. J. Obstet. Gynaecol. 2008, 115, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Charifson, M.A.; Trumble, B.C. Evolutionary origins of polycystic ovary syndrome: An environmental mismatch disorder. Evol. Med. Public Health 2019, 2019, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Azziz, R.; Dumesic, D.A.; Goodarzi, M.O. Polycystic ovary syndrome: An ancient disorder? Fertil. Steril. 2011, 95, 1544–1548. [Google Scholar] [CrossRef] [Green Version]
- Teede, H.; Deeks, H.; Moran, L. Polycystic ovary syndrome: A complex condition with psychological, reproductive and metabolic manifestations that impacts on health across the lifespan. BMC Med. 2010, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Garrido, M.A.; Tena-Sempere, M. Metabolic dysfunction in polycystic ovary syndrome: Pathogenic role of androgen excess and potential therapeutic strategies. Mol. Metab. 2020, 35, 100937. [Google Scholar] [CrossRef]
- Glueck, C.J.; Goldenberg, N. Characteristics of obesity in polycystic ovary syndrome: Etiology, treatment, and genetics. Metabolism 2019, 92, 108–120. [Google Scholar] [CrossRef]
- Reyes-Muñoz, E.; Castellanos-Barroso, G.; Ramírez-Eugenio, B.Y.; Ortega-González, C.; Parra, A.; Castillo-Mora, A.; De La Jara-Díaz, J.F. The risk of gestational diabetes mellitus among Mexican women with a history of infertility and polycystic ovary syndrome. Fertil. Steril. 2012, 97, 1467–1471. [Google Scholar] [CrossRef]
- Rodgers, R.J.; Avery, J.C.; Moore, V.M.; Davies, M.J.; Azziz, R.; Stener-Victorin, E.; Moran, L.J.; Robertson, S.A.; Stepto, N.K.; Norman, R.J.; et al. Complex diseases and co-morbidities: Polycystic ovary syndrome and type 2 diabetes mellitus. Endocr. Connect. 2019, 8, R71–R75. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yao, X.Y.; Shi, R.X.; Liu, S.F.; Wang, X.Y. A potential link between polycystic ovary syndrome and non-alcoholic fatty liver disease: An update meta-analysis. Reprod. Health 2018, 15, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumiceba, V.; López-Cortés, A.; Pérez-Villa, A.; Yumiseba, I.; Guerrero, S.; García-Cárdenas, J.M.; Armendáriz-Castillo, I.; Guevara-Ramírez, P.; Leone, P.E.; Zambrano, A.K.; et al. Oncology and Pharmacogenomics Insights in Polycystic Ovary Syndrome: An Integrative Analysis. Front. Endocrinol. 2020, 11, 840. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hu, J.; Zhang, S.; Fan, W.; Wen, L.; Wang, G.; Zhang, D. Changes in Resting-State Cerebral Activity in Women With Polycystic Ovary Syndrome: A Functional MR Imaging Study. Front. Endocrinol. 2020, 11, 981. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.; Spellacy, W.N.; Prem, K.A.; Cohen, W.D. Hereditary factors in the Stein leventhal 1968. Am. J. Obstet. Gynecol. 1968, 100, 371–387. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Piperi, C. Genetics of polycystic ovary syndrome: Searching for the way out of the labyrinth. Hum. Reprod. Update 2005, 11, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Parker, J. Understanding the Pathogenesis of Polycystic Ovary Syndrome: A transgenerational evolutionary adaptation to lifestyle and the environment. ACNEM J. 2020, 39, 18–26. [Google Scholar]
- Day, F.; Karaderi, T.; Jones, M.R.; Meun, C.; He, C.; Drong, A.; Kraft, P.; Lin, N.; Huang, H.; Broer, L.; et al. Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLoS Genet. 2018, 14, e1007813. [Google Scholar] [CrossRef] [Green Version]
- Crespo, R.P.; Bachega, T.A.S.S.; Mendonça, B.B.; Gomes, L.G. An update of genetic basis of PCOS pathogenesis. Arch. Endocrinol. Metab. 2018, 62, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.R.; Goodarzi, M.O. Genetic determinants of polycystic ovary syndrome: Progress and future directions. Fertil. Steril. 2016, 106, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Varanasi, L.C.; Subasinghe, A.; Jayasinghe, Y.L.; Callegari, E.T.; Garland, S.M.; Gorelik, A.; Wark, J.D. Polycystic ovarian syndrome: Prevalence and impact on the wellbeing of Australian women aged 16–29 years. Aust. N. Z. J. Obstet. Gynaecol. 2018, 58, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Ding, T.; Hardiman, P.J.; Petersen, I.; Wang, F.F.; Qu, F.; Baio, G. The prevalence of polycystic ovary syndrome in reproductiveaged women of different ethnicity: A systematic review and meta-analysis. Oncotarget 2017, 8, 96351–96358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, S.; Zhao, H.; Lu, Z.; Lei, X.; Zhang, Y. Circadian rhythms within the female hpg axis: From physiology to etiology. Endocrinology 2021, 162, bqab117. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xie, N.; Wu, Y.; Zhang, Q.; Zhu, Y.; Dai, M.; Zhou, J.; Pan, J.; Tang, M.; Cheng, Q.; et al. Association between circadian rhythm disruption and polycystic ovary syndrome. Fertil. Steril. 2021, 115, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.J.; Urbanetz, A.A. Environmental toxins and the impact of other endocrine disrupting chemicals in women’s reproductive health. J. Bras. Reprod. Assist. 2019, 23, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.; Chowdhury, O.; Saha, S. Possible link between stress-related factors and altered body composition in women with polycystic ovarian syndrome. J. Hum. Reprod. Sci. 2018, 11, 10–18. [Google Scholar] [CrossRef]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J.; Andersen, M.; Azziz, R.; et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Fertil. Steril. 2018, 110, 364–379. [Google Scholar] [CrossRef] [Green Version]
- Casarini, L.; Simoni, M.; Brigante, G. Is polycystic ovary syndrome a sexual conflict? A review. Reprod. Biomed. Online 2016, 32, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Tremellen, K.; Pearce, K. Dysbiosis of Gut Microbiota [DOGMA]—A novel theory for the development of Polycystic Ovarian Syndrome. Med. Hypotheses 2012, 79, 104–112. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C.; Gersh, F.L. Developmental origins and transgenerational inheritance of polycystic ovary syndrome. Aust. N. Z. J. Obstet. Gynaecol. 2021, 61, 922–926. [Google Scholar] [CrossRef]
- Abbott, D.H.; Dumesic, D.A.; Franks, S. Developmental origin of polycystic ovary syndrome—A hypothesis. J. Endocrinol. 2002, 174, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Rosenfield, R.L.; Ehrmann, D.A. The Pathogenesis of Polycystic Ovary Syndrome [PCOS]: The hypothesis of PCOS as functional ovarian hyperandrogenism revisited. Endocr. Rev. 2016, 37, 467–520. [Google Scholar] [CrossRef]
- Stener-Victorin, E.; Padmanabhan, V.; Walters, K.A.; Campbell, R.E.; Benrick, A.; Giacobini, P.; Dumesic, D.A.; Abbott, D.H. Animal Models to Understand the Etiology and Pathophysiology of Polycystic Ovary Syndrome. Endocr. Rev. 2020, 41, 538–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, D.H.; Dumesic, D.A.; Abbott, D.H. Fetal androgen excess provides a developmental origin for polycystic ovary syndrome. Expert Rev. Obs. Gynecol. 2009, 4, 1–7. [Google Scholar] [CrossRef]
- Abbott, D.H.; Kraynak, M.; Dumesic, D.A.; Levine, J.E. In utero Androgen Excess: A Developmental Commonality Preceding Polycystic Ovary Syndrome? Front. Horm. Res. 2019, 53, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Rayome, B.H.; Dumesic, D.A.; Lewis, K.C.; Edwards, A.K.; Wallen, K.; Wilson, M.E.; Appt, S.E.; Levine, J.E. Clustering of PCOS-like traits in naturally hyperandrogenic female rhesus monkeys. Hum. Reprod. 2017, 32, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, M.; Chow, E.; Aschengrau, A.; Mahalingaiah, S. Prenatal Exposure to Endocrine Disruptors: A Developmental Etiology for Polycystic Ovary Syndrome. Reprod. Sci. 2017, 24, 19–27. [Google Scholar] [CrossRef]
- Neel, J.V. Diabetes Mellitis: A “thrifty” genotype rendered detrimental by “progress”? Am. J. Hum. Genet. 1962, 14, 353–362. [Google Scholar]
- Parker, J.; Hawrelak, J.; Gersh, F.L. Nutritional role of polyphenols as a component of a wholefood diet in the management of polycystic ovary syndrome. J. ACNEM 2021, 40, 6–12. [Google Scholar]
- Tremellen, K.P.K. Nutrition, Fertility, and Human Reproductive Function. In Nutrition, Fertility, and Human Reproductive Function; CRC Press: Adelaide, Astralia, 2015; pp. 27–50. [Google Scholar]
- Rizk, M.G.; Thackray, V.G. Intersection of Polycystic Ovary Syndrome and the Gut Microbiome. J. Endocr. Soc. 2021, 5, bvaa177. [Google Scholar] [CrossRef]
- He, F.F.; Li, Y.M. Role of gut microbiota in the development of insulin resistance and the mechanism underlying polycystic ovary syndrome: A review. J. Ovarian Res. 2020, 13, 73. [Google Scholar] [CrossRef]
- Chen, F. Analysis of the gut microbial composition in polycystic ovary syndrome with acne. Zigong Matern. Child Health Hosp. 2019, 35, 2246–2251. [Google Scholar]
- Zhou, L.; Ni, Z.; Cheng, W.; Yu, J.; Sun, S.; Zhai, D.; Yu, C.; Cai, Z. Characteristic gut microbiota and predicted metabolic functions in women with PCOS. Endocr. Connect. 2020, 9, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, R.; Ostadmohammadi, V.; Akbari, M.; Lankarani, K.B.; Vakili, S.; Peymani, P.; Karamali, M.; Kolahdooz, F.; Asemi, Z. The Effects of Probiotic Supplementation on Clinical Symptom, Weight Loss, Glycemic Control, Lipid and Hormonal Profiles, Biomarkers of Inflammation, and Oxidative Stress in Women with Polycystic Ovary Syndrome: A Systematic Review and Meta-analysis of Ra. Probiotics Antimicrob. Proteins 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Darvishi, S.; Rafraf, M.; Asghari-Jafarabadi, M.; Farzadi, L. Synbiotic Supplementation Improves Metabolic Factors and Obesity Values in Women with Polycystic Ovary Syndrome Independent of Affecting Apelin Levels: A Randomized Double-Blind Placebo-Controlled Clinical Trial. Int. J. Fertil. Steril. 2021, 15, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Karimi, E.; Moini, A.; Yaseri, M.; Shirzad, N.; Sepidarkish, M.; Hossein-Boroujerdi, M.; Hosseinzadeh-Attar, M.J. Effects of synbiotic supplementation on metabolic parameters and apelin in women with polycystic ovary syndrome: A randomised double-blind placebo-controlled trial. Br. J. Nutr. 2018, 119, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Stein, I.F.; Leventhal, M.L. Amenorrhea associated with bilateral polycystic ovaries. Am. J. Obstet. Gynecol. 1935, 29, 181–191. [Google Scholar] [CrossRef]
- Corbett, S.; Morin-Papunen, L. The Polycystic Ovary Syndrome and recent human evolution. Mol. Cell. Endocrinol. 2013, 373, 39–50. [Google Scholar] [CrossRef]
- Rodgers, R.J.; Suturina, L.; Lizneva, D.; Davies, M.J.; Hummitzsch, K.; Irving-Rodgers, H.F.; Robertson, S.A. Is polycystic ovary syndrome a 20th Century phenomenon? Med. Hypotheses 2019, 124, 31–34. [Google Scholar] [CrossRef]
- Holte, J. Polycystic ovary syndrome and insulin resistance: Thrifty genes struggling with over-feeding and sedentary life style? J. Endocrinol. Investig. 1998, 21, 589–601. [Google Scholar] [CrossRef]
- Corbett, S.J.; McMichael, A.J.; Prentice, A.M. Type 2 diabetes, cardiovascular disease, and the evolutionary paradox of the polycystic ovary syndrome: A fertility first hypothesis. Am. J. Hum. Biol. 2009, 21, 587–598. [Google Scholar] [CrossRef]
- Dinsdale, N.L.; Crespi, B.J. Endometriosis and polycystic ovary syndrome are diametric disorders. Evol. Appl. 2021, 14, 1693–1715. [Google Scholar] [CrossRef] [PubMed]
- Sonagra, A.D. Normal Pregnancy—A State of Insulin Resistance. J. Clin. Diagnostic Res. 2014, 8, CC01. [Google Scholar] [CrossRef] [PubMed]
- Lipovka, Y.; Chen, H.; Vagner, J.; Price, T.J.; Tsao, T.S.; Konhilas, J.P. Oestrogen receptors interact with the α-catalytic subunit of AMP-activated protein kinase. Biosci. Rep. 2015, 35, e00264. [Google Scholar] [CrossRef] [Green Version]
- López, M.; Tena-Sempere, M. Estradiol effects on hypothalamic AMPK and BAT thermogenesis: A gateway for obesity treatment? Pharmacol. Ther. 2017, 178, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Rettberg, J.R.; Yao, J.; Brinton, R.D. Estrogen: A master regulator of bioenergetic systems in the brain and body. Front. Neuroendocrinol. 2014, 35, 8–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaaraj, N.; Soliman, A.; Hamed, N.; Alyafei, F.; De Sanctis, V. Understanding the complex role of mtorc as an intracellular critical mediator of whole-body metabolism in anorexia nervosa: A mini review. Acta Biomed. 2021, 92, e2021170. [Google Scholar] [CrossRef] [PubMed]
- Seif, M.W.; Diamond, K.; Nickkho-Amiry, M. Obesity and menstrual disorders. Best Pract. Res. Clin. Obstet. Gynaecol. 2015, 29, 516–527. [Google Scholar] [CrossRef]
- Draper, C.F.; Duisters, K.; Weger, B.; Chakrabarti, A.; Harms, A.C.; Brennan, L.; Hankemeier, T.; Goulet, L.; Konz, T.; Martin, F.P.; et al. Menstrual cycle rhythmicity: Metabolic patterns in healthy women. Sci. Rep. 2018, 8, 14568. [Google Scholar] [CrossRef]
- Roh, E.; Song, D.K.; Kim, M.S. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp. Mol. Med. 2016, 48, e216. [Google Scholar] [CrossRef] [Green Version]
- Ong, Q.; Han, W.; Yang, X. O-GlcNAc as an integrator of signaling pathways. Front. Endocrinol. 2018, 9, 599. [Google Scholar] [CrossRef]
- Gnocchi, D.; Bruscalupi, G. Circadian rhythms and hormonal homeostasis: Pathophysiological implications. Biology 2017, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, D.S.; Aronne, L.J.; Astrup, A.; de Cabo, R.; Cantley, L.C.; Friedman, M.I.; Heymsfield, S.B.; Johnson, J.D.; King, J.C.; Krauss, R.M.; et al. The carbohydrate-insulin model: A physiological perspective on the obesity pandemic. Am. J. Clin. Nutr. 2021, 114, 1873–1885. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A. Developmental and epigenetic pathways to obesity: An evolutionary-developmental perspective. Int. J. Obes. 2008, 32, S62–S71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakumar, P.; Maung-U, K.; Jagadeesh, G. Prevalence and prevention of cardiovascular disease and diabetes mellitus. Pharmacol. Res. 2016, 113, 600–609. [Google Scholar] [CrossRef]
- Crosignani, P.G.; Nicolosi, A.E. Polycystic ovary disease: Heritability and heterogeneity. Hum. Reprod. Update 2001, 7, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vink, J.M.; Sadrzadeh, S.; Lambalk, C.B.; Boomsma, D.I. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J. Clin. Endocrinol. Metab. 2006, 91, 2100–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahsar-Miller, M.D.; Nixon, C.; Boots, L.R.; Go, R.C.; Azziz, R. Prevalence of polycystic ovary syndrome [PCOS] in first-degree relatives of patients with PCOS. Fertil. Steril. 2001, 75, 53–58. [Google Scholar] [CrossRef]
- Dunaif, A. Perspectives in polycystic ovary syndrome: From hair to eternity. J. Clin. Endocrinol. Metab. 2016, 101, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Kosova, G.; Urbanek, M. Genetics of the polycystic ovary syndrome. Mol. Cell. Endocrinol. 2013, 373, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Legro, R.S.; Driscoll, D.; Strauss, J.F.; Fox, J.; Dunaif, A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc. Natl. Acad. Sci. USA 1998, 95, 14956–14960. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitzhugh, W.; et al. Initial sequencing and analysis of the human genome: International Human Genome Sequencing Consortium. Nature 2001, 409, 860–921, Erratum in Nature 2001, 411, 720. [Google Scholar] [CrossRef] [Green Version]
- Belmont, J.W.; Boudreau, A.; Leal, S.M.; Hardenbol, P.; Pasternak, S.; Wheeler, D.A.; Willis, T.D.; Yu, F.; Yang, H.; Gao, Y.; et al. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar] [CrossRef] [Green Version]
- Welt, C.K. Genetics of Polycystic Ovary Syndrome: What is New? Endocrinol. Metab. Clin. N. Am. 2021, 50, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Zhao, H.; He, L.; Shi, Y.; Qin, Y.; Shi, Y.; Li, Z.; You, L.; Zhao, J.; Liu, J.; et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat. Genet. 2011, 43, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.G.; Urbanek, M.; Ehrmann, D.A.; Armstrong, L.L.; Lee, J.Y.; Sisk, R.; Karaderi, T.; Barber, T.M.; McCarthy, M.I.; Franks, S.; et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat. Commun. 2015, 6, 7502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ho, K.; Keaton, J.M.; Hartzel, D.N.; Day, F.; Justice, A.E.; Josyula, N.S.; Pendergrass, S.A.; Actkins, K.E.; Davis, L.K.; et al. A genome-wide association study of polycystic ovary syndrome identified from electronic health records. Am. J. Obstet. Gynecol. 2020, 223, 559.e1–559.e21. [Google Scholar] [CrossRef]
- Zhu, T.; Goodarzi, M.O. Causes and consequences of polycystic ovary syndrome: Insights from Mendelian Randomization. J. Clin. Endocrinol. Metab. 2021. [Google Scholar] [CrossRef]
- Sun, Q.; Gao, Y.; Yang, J.; Lu, J.; Feng, W.; Yang, W. Mendelian Randomization Analysis Identified Potential Genes Pleiotropically Associated with Polycystic Ovary Syndrome. Reprod. Sci. 2021, 1–10. [Google Scholar] [CrossRef]
- Visscher, P.M.; Brown, M.A.; McCarthy, M.I.; Yang, J. Five years of GWAS discovery. Am. J. Hum. Genet. 2012, 90, 7–24. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Rung, J.; Cauchi, S.; Albrechtsen, A.; Shen, L.; Rocheleau, G.; Cavalcanti-Proença, C.; Bacot, F.; Balkau, B.; Belisle, A.; Borch-Johnsen, K.; et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat. Genet. 2009, 41, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Udler, M.S.; McCarthy, M.I.; Florez, J.C.; Mahajan, A. Genetic Risk Scores for Diabetes Diagnosis and Precision Medicine. Endocr. Rev. 2019, 40, 1500–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khera, A.V.; Chaffin, M.; Wade, K.H.; Zahid, S.; Brancale, J.; Xia, R.; Distefano, M.; Senol-Cosar, O.; Haas, M.E.; Bick, A.; et al. Polygenic Prediction of Weight and Obesity Trajectories from Birth to Adulthood. Cell 2019, 177, 587–596.e9. [Google Scholar] [CrossRef] [Green Version]
- Plomin, R.; Haworth, C.M.A.; Davis, O.S.P. Common disorders are quantitative traits. Nat. Rev. Genet. 2009, 10, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Hoyos, L.R.; Chazenbalk, G.D.; Naik, R.; Padmanabhan, V.; Abbott, D.H. Mechanisms of intergenerational transmission of ovary syndrome. Reproduction 2020, 159, R1–R13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloboda, D.M.; Hickey, M.; Hart, R. Reproduction in females: The role of the early life environment. Hum. Reprod. Update 2011, 17, 210–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Li, C.; Liu, X.; Gao, S. Insights into epigenetic patterns in mammalian early embryos. Protein Cell 2021, 12, 7–28. [Google Scholar] [CrossRef] [PubMed]
- Glastras, S.J.; Chen, H.; Pollock, C.A.; Saad, S. Maternal obesity increases the risk of metabolic disease and impacts renal health in offspring. Biosci. Rep. 2018, 38, BSR20180050. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, U.; Armengaud, J.B.; Siddeek, B.; Tolsa, J.F. Perinatal Origins of Adult Disease. Neonatology 2018, 113, 393–399. [Google Scholar] [CrossRef]
- Risnes, K.; Bilsteen, J.F.; Brown, P.; Pulakka, A.; Andersen, A.M.N.; Opdahl, S.; Kajantie, E.; Sandin, S. Mortality Among Young Adults Born Preterm and Early Term in 4 Nordic Nations. JAMA Netw. Open 2021, 4, e2032779. [Google Scholar] [CrossRef]
- Behere, R.V.; Deshmukh, A.S.; Otiv, S.; Gupte, M.D.; Yajnik, C.S. Maternal Vitamin B12 Status During Pregnancy and Its Association With Outcomes of Pregnancy and Health of the Offspring: A Systematic Review and Implications for Policy in India. Front. Endocrinol. 2021, 12, 288. [Google Scholar] [CrossRef] [PubMed]
- Azziz, R. Animal models of pcos not the real thing. Nat. Rev. Endocrinol. 2017, 13, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Poon, K. Behavioral Feeding Circuit: Dietary Fat-Induced Effects of Inflammatory Mediators in the Hypothalamus. Front. Endocrinol. 2020, 11, 905. [Google Scholar] [CrossRef] [PubMed]
- Bateson, P.; Gluckman, P.; Hanson, M. The biology of developmental plasticity and the Predictive Adaptive Response hypothesis. J. Physiol. 2014, 592, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Presley, L.; Minium, J.; Mouzon, S.H. De Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 2009, 32, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, C.S. Transcriptomics and Other Omics Approaches to Investigate Effects of Xenobiotics on the Placenta. Front. Cell Dev. Biol. 2021, 9, 723656. [Google Scholar] [CrossRef]
- De Melo, A.S.; Dias, S.V.; De Carvalho Cavalli, R.; Cardoso, V.C.; Bettiol, H.; Barbieri, M.A.; Ferriani, R.A.; Vieira, C.S. Pathogenesis of polycystic ovary syndrome: Multifactorial assessment from the foetal stage to menopause. Reproduction 2015, 150, R11–R24. [Google Scholar] [CrossRef] [Green Version]
- Schulz, L.C. The Dutch hunger winter and the developmental origins of health and disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16757–16758. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, R. Maternal obesity during pregnancy and cardiovascular development and disease in the offspring. Eur. J. Epidemiol. 2015, 30, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Ishimwe, J.A. Maternal microbiome in preeclampsia pathophysiology and implications on offspring health. Physiol. Rep. 2021, 9, e14875. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.S.B.; Raes, J.; Bork, P. The Human Gut Microbiome: From Association to Modulation. Cell 2018, 172, 1198–1215. [Google Scholar] [CrossRef] [Green Version]
- Davenport, E.R.; Sanders, J.G.; Song, S.J.; Amato, K.R.; Clark, A.G.; Knight, R. The human microbiome in evolution. BMC Biol. 2017, 15, 127. [Google Scholar] [CrossRef]
- Theis, K.R.; Dheilly, N.M.; Klassen, J.L.; Brucker, R.M.; Baines, J.F.; Bosch, T.C.G.; Cryan, J.F.; Gilbert, S.F.; Goodnight, C.J.; Lloyd, E.A.; et al. Getting the Hologenome Concept Right: An Eco-Evolutionary Framework for Hosts and Their Microbiomes. mSystems 2016, 1, e00028-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, A.E.; Werren, J.H. Holes in the hologenome: Why host-microbe symbioses are not holobionts. mBio 2016, 7, e02099-15. [Google Scholar] [CrossRef] [Green Version]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Thingholm, L.B.; Skiecevičie, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.A.; Rühlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in Vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef]
- Liston, A.; Humblet-Baron, S.; Duffy, D.; Goris, A. Human immune diversity: From evolution to modernity. Nat. Immunol. 2021, 22, 1479–1489. [Google Scholar] [CrossRef]
- Meyerson, N.R.; Sawyer, S.L. Two-stepping through time: Mammals and viruses. Trends Microbiol. 2011, 19, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, L.K.; Williams, C.V.; Junge, R.E.; Mahefarisoa, K.L.; Rajaonarivelo, T.; Rakotondrainibe, H.; O’Connell, T.M.; Drea, C.M. A role for gut microbiota in host niche differentiation. ISME J. 2020, 14, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Obin, M.S.; Zhao, L. The gut microbiota, obesity and insulin resistance. Mol. Asp. Med. 2013, 34, 39–58. [Google Scholar] [CrossRef]
- Zhao, X.; Jiang, Y.; Xi, H.; Chen, L.; Feng, X. Exploration of the Relationship between Gut Microbiota and Polycystic Ovary Syndrome [PCOS]: A Review. Geburtshilfe Frauenheilkd. 2020, 80, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Obregon-Tito, A.J.; Tito, R.Y.; Metcalf, J.; Sankaranarayanan, K.; Clemente, J.C.; Ursell, L.K.; Xu, Z.Z.; Treuren, W.; Van Knight, R.; Gaffney, P.M.; et al. Subsistence strategies in traditional societies distinguish gut microbiomes. Nat. Commun. 2015, 6, 6505. [Google Scholar] [CrossRef] [Green Version]
- Smits, S.A.; Leach, J.; Sonnenburg, E.D.; Gonzalez, C.G.; Lichtman, J.S.; Reid, G.; Knight, R.; Manjurano, A.; Changalucha, J.; Elias, J.E.; et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 2017, 357, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Kallus, S.J.; Brandt, L.J. The intestinal microbiota and obesity. J. Clin. Gastroenterol. 2012, 46, 16–24. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [Green Version]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Chatelier, E.; Le Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.L.; Oliveira, F.L.; Azevedo, R. Recent advances in the understanding and management of polycystic ovary syndrome. F1000Research 2019, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Di, N.; Li, L.; Yang, D. Gut microbiota alterations reveal potential gut–brain axis changes in polycystic ovary syndrome. J. Endocrinol. Investig. 2021, 44, 1727–1737. [Google Scholar] [CrossRef]
- Torres, P.J.; Siakowska, M.; Banaszewska, B.; Pawelczyk, L.; Duleba, A.J.; Kelley, S.T.; Thackray, V.G. Gut Microbial Diversity in Women with Polycystic Ovary Syndrome Correlates with Hyperandrogenism. J. Clin. Endocrinol. Metab. 2018, 103, 1502–1511. [Google Scholar] [CrossRef]
- Insenser, M.; Murri, M.; Del Campo, R.; Martínez-García, M.Á.; Fernández-Durán, E.; Escobar-Morreale, H.F. Gut microbiota and the polycystic ovary syndrome: Influence of sex, sex hormones, and obesity. J. Clin. Endocrinol. Metab. 2018, 103, 2552–2562. [Google Scholar] [CrossRef]
- Zhu, X.; Li, Y.; Jiang, Y.; Zhang, J.; Duan, R.; Liu, L.; Liu, C.; Xu, X.; Yu, L.; Wang, Q.; et al. Prediction of Gut Microbial Community Structure and Function in Polycystic Ovary Syndrome With High Low-Density Lipoprotein Cholesterol. Front. Cell Infect. Microbiol. 2021, 11, 665406. [Google Scholar] [CrossRef]
- Lindheim, L.; Bashir, M.; Münzker, J.; Trummer, C.; Zachhuber, V.; Leber, B.; Horvath, A.; Pieber, T.R.; Gorkiewicz, G.; Stadlbauer, V.; et al. Alterations in gut microbiome composition and barrier function are associated with reproductive and metabolic defects in women with polycystic ovary syndrome [PCOS]: A pilot study. PLoS ONE 2017, 12, e0168390. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C.; Hawrelak, J. A narrative review of the role of gastrointestinal dysbiosis in the pathogenesis of polycystic ovary syndrome. Obstet. Gynecol. Sci. 2022, 65, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Xie, W.; Johnson, W.D.; Cefalu, W.T.; Redman, L.M.; Ravussin, E. Defining insulin resistance from hyperinsulinemic-euglycemic clamps. Diabetes Care 2012, 35, 1605–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, C.J.; Prentki, M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diabetes Vasc. Dis. Res. 2019, 16, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Kouli, C.; Alexandraki, K.; Spina, G. Failure of mathematical indices to accurately assess insulin resistance in lean, overweight, or obese women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 1273–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B. Surrogate markers of insulin resistance: A review. World J. Diabetes 2010, 1, 36. [Google Scholar] [CrossRef]
- Wu, X.K.; Zhou, S.Y.; Liu, J.X.; Pöllänen, P.; Sallinen, K.; Mäkinen, M.; Erkkola, R. Selective ovary resistance to insulin signaling in women with polycystic ovary syndrome. Fertil. Steril. 2003, 80, 954–965. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus Total Insulin Resistance: A Pathogenic Paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Hardy, O.T.; Czech, M.P.; Corvera, S. What causes the insulin resistance underlying obesity? Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Toosy, S.; Sodi, R.; Pappachan, J.M. Lean polycystic ovary syndrome [PCOS]: An evidence-based practical approach. J. Diabetes Metab. Disord. 2018, 17, 277–285. [Google Scholar] [CrossRef]
- Wang, J.; Wu, D.; Guo, H.; Li, M. Hyperandrogenemia and insulin resistance: The chief culprit of polycystic ovary syndrome. Life Sci. 2019, 236. [Google Scholar] [CrossRef]
- Cibula, D. Is insulin resistance an essential component of PCOS? The influence of confounding factors. Hum. Reprod. 2004, 19, 757–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassar, S.; Misso, M.L.; Hopkins, W.G.; Shaw, C.S.; Teede, H.J.; Stepto, N.K. Insulin resistance in polycystic ovary syndrome: A systematic review and meta-analysis of euglycaemic-hyperinsulinaemic clamp studies. Hum. Reprod. 2016, 31, 2619–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepto, N.K.; Cassar, S.; Joham, A.E.; Hutchison, S.K.; Harrison, C.L.; Goldstein, R.F.; Teede, H.J. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic-hyperinsulaemic clamp. Hum. Reprod. 2013, 28, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Rubin, K.H.; Glintborg, D.; Nybo, M.; Abrahamsen, B.; Andersen, M. Development and risk factors of type 2 diabetes in a nationwide population of women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 3848–3857. [Google Scholar] [CrossRef] [Green Version]
- Crofts, C.A.P. Hyperinsulinemia: A unifying theory of chronic disease? Diabesity 2015, 1, 34. [Google Scholar] [CrossRef] [Green Version]
- Watve, M.G.; Yajnik, C.S. Evolutionary origins of insulin resistance: A behavioral switch hypothesis. BMC Evol. Biol. 2007, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Khalid, M.; Alkaabi, J.; Khan, M.A.B.; Adem, A. Insulin signal transduction perturbations in insulin resistance. Int. J. Mol. Sci. 2021, 22, 8590. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Rosa, S.C.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 2020, 8, e14607. [Google Scholar] [CrossRef]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef] [Green Version]
- Soeters, M.R.; Soeters, P.B.; Schooneman, M.G.; Houten, S.M.; Romijn, J.A. Adaptive reciprocity of lipid and glucose metabolism in human short-term starvation. Am. J. Physiol.-Endocrinol. Metab. 2012, 303, 1397–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, X.; Zhou, W.; Messina, J.L. Acute psychological stress results in the rapid development of insulin resistance. J. Endocrinol. 2013, 217, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morciano, A.; Romani, F.; Sagnella, F.; Scarinci, E.; Palla, C.; Moro, F.; Tropea, A.; Policola, C.; Della Casa, S.; Guido, M.; et al. Assessment of insulin resistance in lean women with polycystic ovary syndrome. Fertil. Steril. 2014, 102, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Small, L.; Brandon, A.E.; Turner, N.; Cooney, G.J. Modeling insulin resistance in rodents by alterations in diet: What have high-fat and high-calorie diets revealed? Am. J. Physiol.-Endocrinol. Metab. 2018, 314, E251–E265. [Google Scholar] [CrossRef]
- Hallberg, S.J.; Gershuni, V.M.; Hazbun, T.L.; Athinarayanan, S.J. Reversing Type 2 Diabetes: A Narrative Review of the Evidence. Nutrients 2019, 11, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, N.; Xue, M.; Weickert, M.; Thornally, P. Reversal of Insulin Resistance in Overweight and Obese Subjects by trans-Resveratrol and Hesperetin Combination-Link to Dysglycemia, Blood Pressure, Dyslipidemia, and Low-Grade Inflammation. Nutrients 2021, 13, 2374. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Zhou, H.; Hu, M.; Feng, H. Effect of diet on insulin resistance in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2020, 105, 3346–3360. [Google Scholar] [CrossRef]
- Kazemi, A.; Soltani, S.; Ghorabi, S.; Keshtkar, A.; Daneshzad, E.; Nasri, F.; Mazloomi, S.M. Effect of probiotic and synbiotic supplementation on inflammatory markers in health and disease status: A systematic review and meta-analysis of clinical trials. Clin. Nutr. 2020, 39, 789–819. [Google Scholar] [CrossRef]
- Street, M.E.; Bernasconi, S. Endocrine-disrupting chemicals in human fetal growth. Int. J. Mol. Sci. 2020, 21, 1430. [Google Scholar] [CrossRef] [Green Version]
- Patterson, R.E.; Laughlin, G.A.; LaCroix, A.Z.; Hartman, S.J.; Natarajan, L.; Senger, C.M.; Martínez, M.E.; Villaseñor, A.; Sears, D.D.; Marinac, C.R.; et al. Intermittent Fasting and Human Metabolic Health. J. Acad. Nutr. Diet. 2015, 115, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Adeva-Andany, M.M.; González-Lucán, M.; Fernández-Fernández, C.; Carneiro-Freire, N.; Seco-Filgueira, M.; Pedre-Piñeiro, A.M. Effect of diet composition on insulin sensitivity in humans. Clin. Nutr. ESPEN 2019, 33, 29–38. [Google Scholar] [CrossRef] [PubMed]
- González, F.; Considine, R.V.; Abdelhadi, O.A.; Xue, J.; Acton, A.J. Saturated fat ingestion stimulates proatherogenic inflammation in polycystic ovary syndrome. Am. J. Physiol. Metab. 2021, 321, E689–E701. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, E.; Malagoli, D.; Franceschi, C. The evolution of the adipose tissue: A neglected enigma. Gen. Comp. Endocrinol. 2011, 174, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Müller-Wieland, D.; Kotzka, J. Lipodystrophies—disorders of the fatty tissue. Int. J. Mol. Sci. 2020, 21, 8778. [Google Scholar] [CrossRef]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Pawłowski, B.; Żelaźniewicz, A. The evolution of perennially enlarged breasts in women: A critical review and a novel hypothesis. Biol. Rev. 2021, 2798, 2794–2809. [Google Scholar] [CrossRef]
- McLaughlin, T.; Lamendola, C.; Liu, A.; Abbasi, F. Preferential fat deposition in subcutaneous versus visceral depots is associated with insulin sensitivity. J. Clin. Endocrinol. Metab. 2011, 96, 1756–1760. [Google Scholar] [CrossRef] [Green Version]
- Barrea, L.; Frias-Toral, E.; Verde, L.; Ceriani, F.; Cucalón, G.; Garcia-Velasquez, E.; Moretti, D.; Savastano, S.; Colao, A.; Muscogiuri, G. PCOS and nutritional approaches: Differences between lean and obese phenotype. Metab. Open 2021, 12, 100123. [Google Scholar] [CrossRef]
- Sellayah, D. The impact of early human migration on brown adipose tissue evolution and its relevance to the modern obesity pandemic. J. Endocr. Soc. 2019, 3, 372–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchard, C.; Tremblay, A.; Després, J.P.; Nadeau, A.; Lupien, P.J.; Thériault, G.; Dussault, J.; Moorjani, S.; Pinault, S.; Fournier, G. The response to long-term overfeeding in identical twins. N. Engl. J. Med. 1990, 322, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.M.; Hanson, P.; Weickert, M.O.; Franks, S. Obesity and Polycystic Ovary Syndrome: Implications for Pathogenesis and Novel Management Strategies. Clin. Med. Insights Reprod. Health 2019, 13, 1179558119874042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, I.; Garg, A. Lipodystrophy Syndromes. Endocrinol. Metab. Clin. N. Am. 2016, 45, 783–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambineri, A.; Zanotti, L. Polycystic ovary syndrome in familial partial lipodystrophy type 2 [Fpld2]: Basic and clinical aspects. Nucleus 2018, 9, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy [Dunnigan variety]. J. Clin. Endocrinol. Metab. 2000, 85, 1776–1782. [Google Scholar] [CrossRef]
- Kahn, L.G.; Philippat, C.; Nakayama, S.F.; Slama, R.; Trasande, L. Endocrine-disrupting chemicals: Implications for human health. Lancet Diabetes Endocrinol. 2020, 8, 703–718. [Google Scholar] [CrossRef]
- Whitmee, S.; Haines, A.; Beyrer, C.; Boltz, F.; Capon, A.G.; De Souza Dias, B.F.; Ezeh, A.; Frumkin, H.; Gong, P.; Head, P.; et al. Safeguarding human health in the Anthropocene epoch: Report of the Rockefeller Foundation-Lancet Commission on planetary health. Lancet 2015, 386, 1973–2028. [Google Scholar] [CrossRef]
- Schug, T.T.; Johnson, A.F.; Birnbaum, L.S.; Colborn, T.; Guillette, L.J.; Crews, D.P.; Collins, T.; Soto, A.M.; Vom Saal, F.S.; McLachlan, J.A.; et al. Minireview: Endocrine disruptors: Past lessons and future directions. Mol. Endocrinol. 2016, 30, 833–847. [Google Scholar] [CrossRef] [Green Version]
- TEDX List of Potential Endocrine Disruptors the Endocrine Disruptor Exchange. 2018. Available online: https://endocrinedisruption.org/interactive-tools/tedx-list-of-potential-endocrine-disruptors/search-the-tedx-list (accessed on 15 November 2021).
- Eskenazi, B.; Chevrier, J.; Rauch, S.A.; Kogut, K.; Harley, K.G.; Johnson, C.; Trujillo, C.; Sjödin, A.; Bradman, A. In utero and childhood polybrominated diphenyl ether [PBDE] exposures and neurodevelopment in the CHAMACOS study. Environ. Health Perspect. 2013, 121, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Parker, J. Glyphosate induced intestinal permeability in the pathogenesis of PCOS. ACNEM J. 2015, 34, 3–7. [Google Scholar]
- Mitro, S.D.; Johnson, T.; Zota, A.R. Cumulative Chemical Exposures During Pregnancy and Early Development. Curr. Environ. Health Rep. 2015, 2, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, D.B.; Bishop, A.; Needham, L.L. Concentrations of xenobiotic chemicals in the maternal-fetal unit. Reprod. Toxicol. 2007, 23, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Starling, A.P.; Adgate, J.L.; Hamman, R.F.; Kechris, K.; Calafat, A.M.; Ye, X.; Dabelea, D. Perfluoroalkyl substances during pregnancy and offspring weight and adiposity at birth: Examining mediation by maternal fasting glucose in the healthy start study. Environ. Health Perspect. 2017, 125, 067016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohajer, N.; Du, C.Y.; Checkcinco, C.; Blumberg, B. Obesogens: How They Are Identi fied and Molecular Mechanisms Underlying Their Action. Front. Endocrinol. 2021, 12, 1503. [Google Scholar] [CrossRef] [PubMed]
- De Araújo, J.F.P.; Podratz, P.L.; Sena, G.C.; Merlo, E.; Freitas-Lima, L.C.; Ayub, J.G.M.; Pereira, A.F.Z.; Santos-Silva, A.P.; Miranda-Alves, L.; Silva, I.V.; et al. The obesogen tributyltin induces abnormal ovarian adipogenesis in adult female rats. Toxicol. Lett. 2018, 295, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Dumesic, D.A.; Levine, J.E.; Angeles, L. Hyperandrogenic origins of polycystic ovary syndrome-Implications for Pathophysiology and Therapy. Expert Rev. Endocrinol. Metab. 2019, 14, 131–143. [Google Scholar] [CrossRef]
- Resnik, D.B. The precautionary principle and medical decision making. J. Med. Philos. 2004, 29, 281–299. [Google Scholar] [CrossRef]
- Di Renzo, G.C.; Conry, J.A.; Blake, J.; Defrancesco, M.S.; Denicola, N.; Martin, J.N.; McCue, K.A.; Richmond, D.; Shah, A.; Sutton, P.; et al. International Federation of Gynecology and Obstetrics opinion on reproductive health impacts of exposure to toxic environmental chemicals. Int. J. Gynecol. Obstet. 2015, 131, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef]
- Chemical Exposures During Pregnancy: Dealing with Potential, but Unproven, Risks to Child Health. Available online: https://eprints.gla.ac.uk/97765/ (accessed on 3 August 2021).
- Shishehgar, F.; Ramezani Tehrani, F.; Mirmiran, P.; Hajian, S.; Baghestani, A.R.; Moslehi, N. Comparison of Dietary Intake between Polycystic Ovary Syndrome Women and Controls. Glob. J. Health Sci. 2016, 8, 302. [Google Scholar] [CrossRef] [PubMed]
- Rajska, A.; Buszewska-Forajta, M.; Rachoń, D.; Markuszewski, M.J. Metabolomic insight into polycystic ovary syndrome—An overview. Int. J. Mol. Sci. 2020, 21, 4853. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.S.; Hutchison, S.K.; Van Ryswyk, E.; Norman, R.J.; Teede, H.J.; Moran, L.J. Lifestyle changes in women with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, N.; Geelen, A.; Streppel, M.T.; De Groot, L.C.P.G.M.; Orfanos, P.; Van Den Hooven, E.H.; Pikhart, H.; Boffetta, P.; Trichopoulou, A.; Bobak, M.; et al. Adherence to a healthy diet according to the world health organization guidelines and all-cause mortality in elderly adults from Europe and the United States. Am. J. Epidemiol. 2014, 180, 978–988. [Google Scholar] [CrossRef]
- Kim, H.; Rebholz, C.M. Metabolomic biomarkers of healthy dietary patterns and cardiovascular outcomes. Curr. Atheroscler. Rep. 2021, 23, 26. [Google Scholar] [CrossRef] [PubMed]
- Pontzer, H.; Wood, B.M.; Raichlen, D.A. Hunter-gatherers as models in public health. Obes. Rev. 2018, 19, 24–35. [Google Scholar] [CrossRef]
- Fayet-Moore, F.; Cassettari, T.; Tuck, K.; McConnell, A.; Petocz, P. Dietary fibre intake in australia. Paper i: Associations with demographic, socio-economic, and anthropometric factors. Nutrients 2018, 10, 599. [Google Scholar] [CrossRef] [Green Version]
- Satija, A.; Hu, F.B. Cardiovascular benefits of dietary fiber. Curr. Atheroscler. Rep. 2012, 14, 505–514. [Google Scholar] [CrossRef]
- Müller, M.; Canfora, E.E.; Blaak, E.E. Gastrointestinal transit time, glucose homeostasis and metabolic health: Modulation by dietary fibers. Nutrients 2018, 10, 275. [Google Scholar] [CrossRef] [Green Version]
- Lattimer, J.M.; Haub, M.D. Effects of dietary fiber and its components on metabolic health. Nutrients 2010, 2, 1266–1289. [Google Scholar] [CrossRef] [Green Version]
- Ghanim, H.; Batra, M.; Abuaysheh, S.; Green, K.; Makdissi, A.; Kuhadiya, N.D.; Chaudhuri, A.; Dandona, P. Antiinflammatory and ROS Suppressive Effects of the Addition of Fiber to a High-Fat High-Calorie Meal. J. Clin. Endocrinol. Metab. 2017, 102, 858–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, S.V.; Hannon, B.A.; An, R.; Holscher, H.D. Effects of isolated soluble fiber supplementation on body weight, glycemia, and insulinemia in adults with overweight and obesity: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2017, 106, 1514–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veronese, N.; Solmi, M.; Caruso, M.G.; Giannelli, G.; Osella, A.R.; Evangelou, E.; Maggi, S.; Fontana, L.; Stubbs, B.; Tzoulaki, I. Dietary fiber and health outcomes: An umbrella review of systematic reviews and meta-analyses. Am. J. Clin. Nutr. 2018, 107, 436–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, D.A.; Pride, S.M.; Cheung, A.P. Low intakes of dietary fiber and magnesium are associated with insulin resistance and hyperandrogenism in polycystic ovary syndrome: A cohort study. Food Sci. Nutr. 2019, 7, 1426–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaix, A.; Zarrinpar, A.; Panda, S. The circadian coordination of cell biology. J. Cell Biol. 2016, 215, 15–25. [Google Scholar] [CrossRef]
- Bhadra, U.; Thakkar, N.; Das, P.; Pal Bhadra, M. Evolution of circadian rhythms: From bacteria to human. Sleep Med. 2017, 35, 49–61. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Lazar, M.A. Circadian metabolism in the light of evolution. Endocr. Rev. 2015, 36, 289–304. [Google Scholar] [CrossRef]
- Palm, D.; Uzoni, A.; Simon, F.; Fischer, M.; Coogan, A.; Tucha, O.; Thome, J.; Faltraco, F. Evolutionary conservations, changes of circadian rhythms and their effect on circadian disturbances and therapeutic approaches. Neurosci. Biobehav. Rev. 2021, 128, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.; O’Neill, J.S.; Maywood, E.S. Circadian clocks: Regulators of endocrine and metabolic rhythms. J. Endocrinol. 2007, 195, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Rosenwasser, A.M.; Turek, F.W. Neurobiology of circadian rhythm regulation. Sleep Med. Clin. 2015, 10, 403–412. [Google Scholar] [CrossRef]
- Lucas, J.A.; Schmidt, T.M. Cellular properties of intrinsically photosensitive retinal ganglion cells during postnatal development. Neural Dev. 2019, 14, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, G.D.M.; Cade, J.E.; Grant, P.J.; Hardie, L.J. Nutrition and the circadian system. Br. J. Nutr. 2016, 116, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patke, A.; Young, M.W.; Axelrod, S. Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 2020, 21, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Voigt, R.M.; Forsyth, C.B.; Green, S.J.; Engen, P.A.; Keshavarzian, A. Circadian Rhythm and the Gut Microbiome, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 131. [Google Scholar]
- Julius, A.A.; Yin, J.; Wen, J.T. Time optimal entrainment control for circadian rhythm. PLoS ONE 2019, 14, e0225988. [Google Scholar] [CrossRef] [PubMed]
- Bass, J.; Takahashi, J.S. Circadian integration of metabolism and energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Milanova, I.V.; Kalsbeek, M.J.T.; Wang, X.L.; Korpel, N.L.; Stenvers, D.J.; Wolff, S.E.C.; De Goede, P.; Heijboer, A.C.; Fliers, E.; La Fleur, S.E.; et al. Diet-induced obesity disturbs microglial immunometabolism in a time-of-day manner. Front. Endocrinol. 2019, 10, 424. [Google Scholar] [CrossRef]
- Jakubowicz, D.; Barnea, M.; Wainstein, J.; Froy, O. High Caloric intake at breakfast vs. dinner differentially influences weight loss of overweight and obese women. Obesity 2013, 21, 2504–2512. [Google Scholar] [CrossRef]
- Oike, H.; Oishi, K.; Kobori, M. Nutrients, Clock Genes, and Chrononutrition. Curr. Nutr. Rep. 2014, 3, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, B.M.; Zierath, J.R. Circadian rhythms and exercise—Re-setting the clock in metabolic disease. Nat. Rev. Endocrinol. 2019, 15, 197–206. [Google Scholar] [CrossRef]
- Sridhar, G.R.; Sanjana, N.S.N. Sleep, circadian dysrhythmia, obesity and diabetes. World J. Diabetes 2016, 7, 515. [Google Scholar] [CrossRef]
- Ganesan, S.; Magee, M.; Stone, J.E.; Mulhall, M.D.; Collins, A.; Howard, M.E.; Lockley, S.W.; Rajaratnam, S.M.W.; Sletten, T.L. The Impact of Shift Work on Sleep, Alertness and Performance in Healthcare Workers. Sci. Rep. 2019, 9, 4635. [Google Scholar] [CrossRef] [PubMed]
- Kopp, R.; Martínez, I.O.; Legradi, J.; Legler, J. Exposure to endocrine disrupting chemicals perturbs lipid metabolism and circadian rhythms. J. Environ. Sci. 2017, 62, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Tahara, Y.; Aoyama, S.; Shibata, S. The mammalian circadian clock and its entrainment by stress and exercise. J. Physiol. Sci. 2017, 67, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papalou, O.; Diamanti-Kandarakis, E. The role of stress in PCOS. Expert Rev. Endocrinol. Metab. 2017, 12, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.L.; McWhirter, L.; Diniz Behn, C.; Bubar, K.M.; Kaar, J.L.; Pyle, L.; Rahat, H.; Garcia-Reyes, Y.; Carreau, A.M.; Wright, K.P.; et al. Morning Circadian Misalignment Is Associated with Insulin Resistance in Girls with Obesity and Polycystic Ovarian Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 3525–3534. [Google Scholar] [CrossRef]
- Zhou, X.; Huddleston, H. Let there be light: Does circadian rhythm disruption cause polycystic ovary syndrome? Fertil. Steril. 2021, 115, 607–608. [Google Scholar] [CrossRef]
- Amaral, F.G.; Castrucci, A.M.; Cipolla-Neto, J.; Poletini, M.O.; Mendez, N.; Richter, H.G.; Sellix, M.T. Environmental control of biological rhythms: Effects on development, fertility and metabolism. J. Neuroendocrinol. 2014, 26, 603–612. [Google Scholar] [CrossRef]
- Gurusinghe, D.; Gill, S.; Almario, R.U.; Lee, J.; Horn, W.F.; Keim, N.L.; Kim, K.; Karakas, S.E. In polycystic ovary syndrome, adrenal steroids are regulated differently in the morning versus in response to nutrient intake. Fertil. Steril. 2010, 93, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Roelfsema, F.; Kok, P.; Pereira, A.M.; Pijl, H. Cortisol production rate is similarly elevated in obese women with or without the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 3318–3324. [Google Scholar] [CrossRef] [Green Version]
- Bravo, R.; Ugartemendia, L.; Cubero, J. Current Opinions in Chrononutrition and Health. J. Clin. Nutr. Diet. 2017, 3, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Adafer, R.; Messaadi, W.; Meddahi, M.; Patey, A.; Haderbache, A.; Bayen, S.; Messaadi, N. Food Timing, Circadian Rhythm and Chrononutrition: A Systematic Review of Time-Restricted Eating’ s Effects on Human Health. Nutrients 2020, 12, 3770. [Google Scholar] [CrossRef] [PubMed]
- Challet, E.; Kalsbeek, A. Editorial: Circadian rhythms and metabolism. Front. Endocrinol. 2017, 8, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamshed, H.; Beyl, R.A.; Manna Della, D.L.; Yang, E.S.; Ravussin, E.; Peterson, C.M. Early Time-Restricted Feeding Improves 24-Hour glucose levels and affects markers of the circadian clock, aging, and autophagy in humans. Nutrients 2019, 11, 1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touitou, Y.; Touitou, D.; Reinberg, A. Disruption of adolescents’ circadian clock: The vicious circle of media use, exposure to light at night, sleep loss and risk behaviors. J. Physiol. Paris 2016, 110, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Tähkämö, L.; Partonen, T.; Pesonen, A.K. Systematic review of light exposure impact on human circadian rhythm. Chronobiol. Int. 2019, 36, 151–170. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, J.; O’Brien, C.; Hawrelak, J.; Gersh, F.L. Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. Int. J. Environ. Res. Public Health 2022, 19, 1336. https://doi.org/10.3390/ijerph19031336
Parker J, O’Brien C, Hawrelak J, Gersh FL. Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. International Journal of Environmental Research and Public Health. 2022; 19(3):1336. https://doi.org/10.3390/ijerph19031336
Chicago/Turabian StyleParker, Jim, Claire O’Brien, Jason Hawrelak, and Felice L. Gersh. 2022. "Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment" International Journal of Environmental Research and Public Health 19, no. 3: 1336. https://doi.org/10.3390/ijerph19031336
APA StyleParker, J., O’Brien, C., Hawrelak, J., & Gersh, F. L. (2022). Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. International Journal of Environmental Research and Public Health, 19(3), 1336. https://doi.org/10.3390/ijerph19031336