
Can Exposure to Environmental Pollutants Be Associated with Less Effective Chemotherapy in Cancer Patients?


Abstract
:1. Introduction
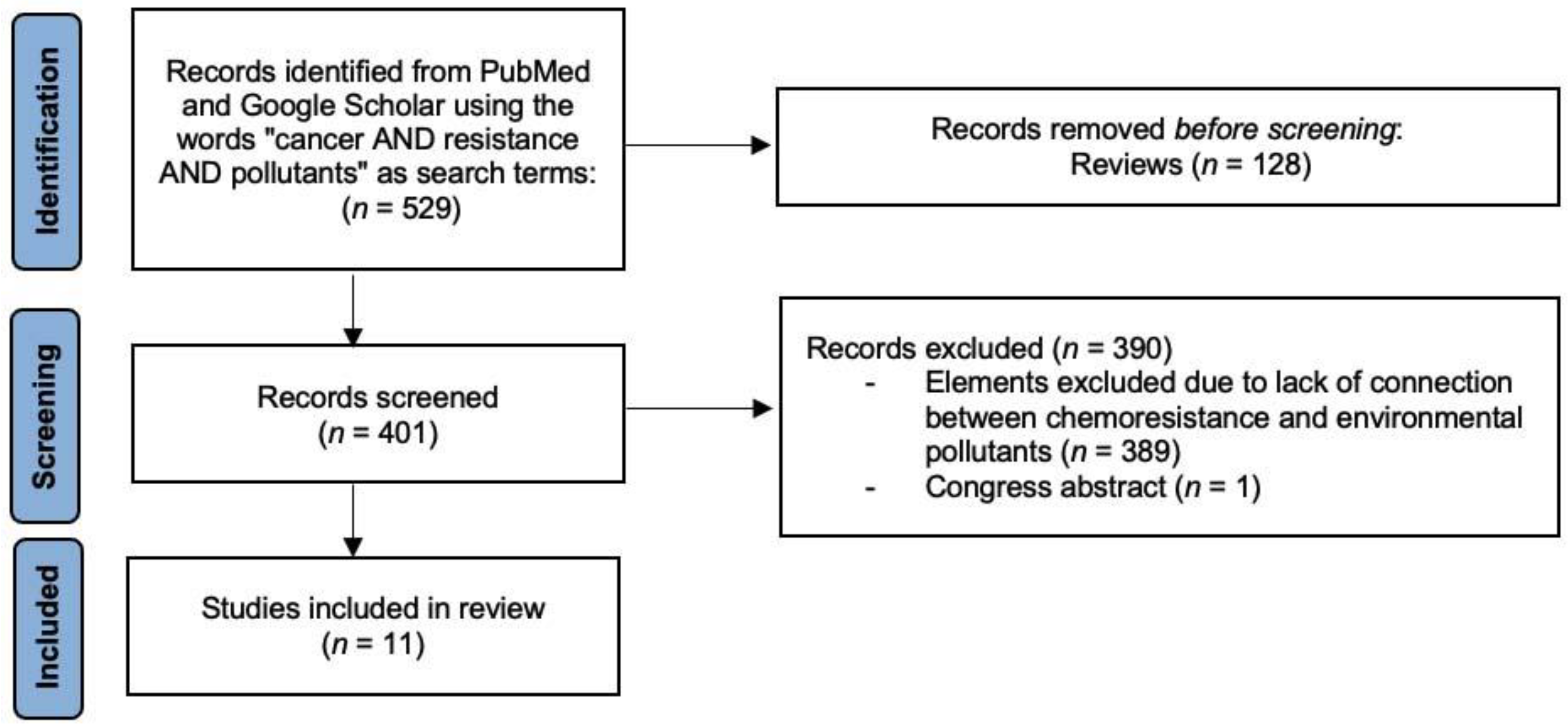
2. Methods
3. Mechanisms of Action of Chemotherapy Drugs
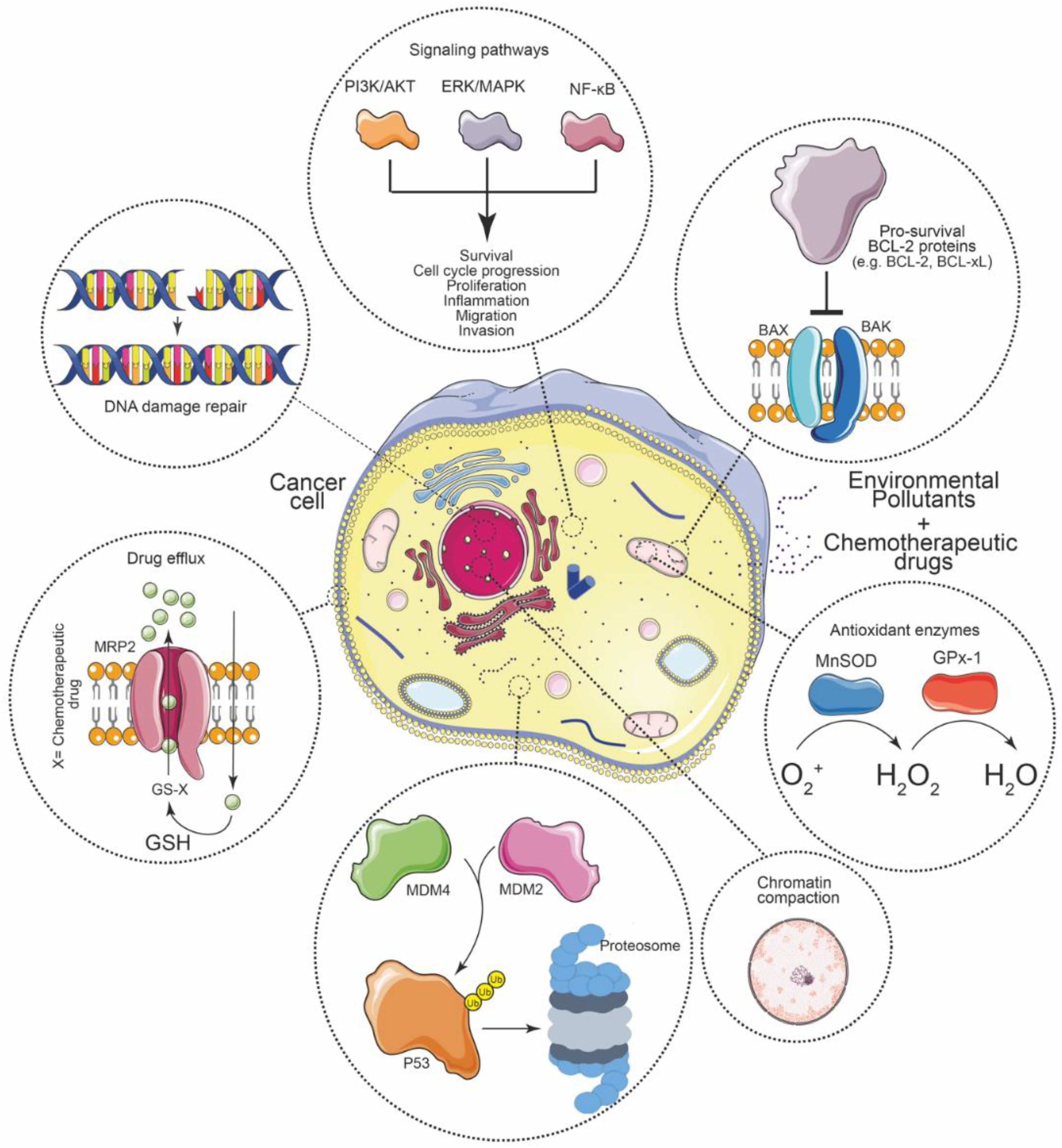
4. Molecular Mechanisms of Chemoresistance in Cancer
4.1. Enhanced Drug Efflux
4.2. Activation of Oncogenic Signaling Pathways
4.3. Increased DNA Repair Capacity
4.4. Elevated Xenobiotic Metabolism
4.5. Increased CSCs
4.6. Extracellular Matrix (ECM)
5. Evidence of Pollutants Affecting the Efficacy of Chemotherapeutic Drugs
5.1. Bisphenol A (BPA)
5.2. Benzo[a]pyrene (BaP)
5.3. Persistent Organic Pollutants (POPs)
5.4. Aluminum Chloride (AlCl3)
5.5. Airborne Particulate Matter (PM)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DN(M)EL | Derived no-effect or minimum effect level |
| BPA | Bisphenol A |
| POP | Persistent organic pollutant |
| BaP | Benzo[a]pyrene |
| AlCl3 | Aluminum chloride |
| PCB-1254 | Polychlorinated biphenyls |
| HBCD | Hexabromocyclododecane |
| OP | 4-tert-octylphenol |
| DEHP | Bis (2-ethylhexyl) phthalate |
| PAH | Polycyclic aromatic hydrocarbon |
| CDDP | Cisplatin |
| DOX | Doxorubicin |
| VIN | Vinblastine |
| 5-FU | 5-fluorouracil |
| PTX | Paclitaxel |
| CPT | Camptothecin |
| HCC | Hepatocellular carcinoma |
| BC | Breast cancer |
| COAD | Colon adenocarcinoma |
| OC | Oesophageal cancer |
| SCC | Squamous cell carcinoma |
| MEF | Mouse embryonic fibroblasts |
| CNM | Concentration not mentioned |
| EMT | Epithelial-to-mesenchymal transition |
| CSC | Cancer stem cell |
| ABC | ATP-binding cassette |
| QUIN | Quinidine |
| VP-16 | Etoposide |
| MTX | Methotrexate |
| TMZ | Temozolomide |
| FLUT | Flutamide |
| TXT | Docetaxel |
| IFO | Ifosfamide |
| CTX | Cyclophosphamide |
| VIN | Vinblastine |
| GSH | Glutathione |
| NER | Nucleotide excision repair |
| HR | Homologous recombination |
| MGMT | O6-methylguanine-DNA methyltransferase |
| TLS | Translesion synthesis |
| OCP | Organochlorine pesticides |
| PBDE | Polybrominated diphenyl ethers |
| PFOS | Perfluorooctane sulfonate |
| PCDD | Polychlorinated dibenzo-p-dioxins |
| PCDF | Polychlorinated dibenzofurans |
| HBCD | Hexabromocyclododecane |
References
- Landrigan, P.J.; Fuller, R.; Acosta, N.J.R.; Adeyi, O.; Arnold, R.; Basu, N.; Baldé, A.B.; Bertollini, R.; Bose-O’Reilly, S.; Boufford, J.I.; et al. The Lancet Commission on pollution and health. Lancet 2018, 391, 462–512. [Google Scholar] [CrossRef] [Green Version]
- Iqubal, A.; Ahmed, M.; Ahmad, S.; Sahoo, C.R.; Iqubal, M.K.; Haque, S.E. Environmental neurotoxic pollutants: Review. Environ. Sci. Pollut. Res. 2020, 27, 41175–41198. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.; Nawrot, T.S.; Baccarelli, A.A. Hallmarks of environmental insults. Cell 2021, 184, 1455–1468. [Google Scholar] [CrossRef] [PubMed]
- Goodson, W.H.; Lowe, L.; Carpenter, D.O.; Gilbertson, M.; Manaf Ali, A.; Lopez de Cerain Salsamendi, A.; Lasfar, A.; Carnero, A.; Azqueta, A.; Amedei, A.; et al. Assessing the carcinogenic potential of low-dose exposures to chemical mixtures in the environment: The challenge ahead. Carcinogenesis 2015, 36, S254–S296. [Google Scholar] [CrossRef] [Green Version]
- Ochieng, J.; Nangami, G.N.; Ogunkua, O.; Miousse, I.R.; Koturbash, I.; Odero-Marah, V.; McCawley, L.J.; Nangia-Makker, P.; Ahmed, N.; Luqmani, Y.; et al. The impact of low-dose carcinogens and environmental disruptors on tissue invasion and metastasis. Carcinogenesis 2015, 36, S128–S159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Urrego, D.; Rodríguez-Urrego, L. Air quality during the COVID-19: PM2.5 analysis in the 50 most polluted capital cities in the world. Environ. Pollut. 2020, 266, 115042. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Santibáñez-Andrade, M.; Chirino, Y.I.; González-Ramírez, I.; Sánchez-Pérez, Y.; García-Cuellar, C.M. Deciphering the Code between Air Pollution and Disease: The Effect of Particulate Matter on Cancer Hallmarks. Int. J. Mol. Sci. 2019, 21, 136. [Google Scholar] [CrossRef] [Green Version]
- Boffetta, P. Human cancer from environmental pollutants: The epidemiological evidence. Mutat. Res. Toxicol. Environ. Mutagen. 2006, 608, 157–162. [Google Scholar] [CrossRef]
- Koual, M.; Tomkiewicz, C.; Cano-Sancho, G.; Antignac, J.-P.; Bats, A.-S.; Coumoul, X. Environmental chemicals, breast cancer progression and drug resistance. Environ. Health 2020, 19, 117. [Google Scholar] [CrossRef]
- Yeldag, G.; Rice, A.; Del Río Hernández, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPensee, E.W.; Tuttle, T.R.; Fox, S.R.; Ben-Jonathan, N. Bisphenol A at Low Nanomolar Doses Confers Chemoresistance in Estrogen Receptor-α–Positive and –Negative Breast Cancer Cells. Environ. Health Perspect. 2009, 117, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPensee, E.W.; LaPensee, C.R.; Fox, S.; Schwemberger, S.; Afton, S.; Ben-Jonathan, N. Bisphenol A and estradiol are equipotent in antagonizing cisplatin-induced cytotoxicity in breast cancer cells. Cancer Lett. 2010, 290, 167–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, M.; Ribeiro-Varandas, E. Bisphenol A at the reference level counteracts doxorubicin transcriptional effects on cancer related genes in HT29 cells. Toxicol. Vitr. 2015, 29, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Ladeira, C.; Zeferino, S.; Dias, A.; Faria, I.; Cristovam, E.; Gomes, M.; Ribeiro, E. Cytotoxic and genotoxic effects of environmental relevant concentrations of bisphenol A and interactions with doxorubicin. Mutat. Res. Toxicol. Environ. Mutagen. 2019, 838, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Sonavane, M.; Sykora, P.; Andrews, J.F.; Sobol, R.W.; Gassman, N.R. Camptothecin Efficacy to Poison Top1 Is Altered by Bisphenol A in Mouse Embryonic Fibroblasts. Chem. Res. Toxicol. 2018, 31, 510–519. [Google Scholar] [CrossRef]
- Dzobo, K.; Hassen, N.; Senthebane, D.; Thomford, N.; Rowe, A.; Shipanga, H.; Wonkam, A.; Parker, M.; Mowla, S.; Dandara, C. Chemoresistance to Cancer Treatment: Benzo-α-Pyrene as Friend or Foe? Molecules 2018, 23, 930. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Xiao, X.; Yao, Y.; Yu, J.; Chen, Q.; Liang, P.; Zhang, Y. Benzo[a]pyrene promotes progression in tongue squamous cell carcinoma. Oral Dis. 2020, 26, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cui, Z.; Zakki, S.A.; Feng, Q.; Sun, L.; Feril, L.B.; Inadera, H. Aluminum chloride causes 5-fluorouracil resistance in hepatocellular carcinoma HepG2 cells. J. Cell. Physiol. 2019, 234, 20249–20265. [Google Scholar] [CrossRef]
- Merk, R.; Heßelbach, K.; Osipova, A.; Popadić, D.; Schmidt-Heck, W.; Kim, G.-J.; Günther, S.; Piñeres, A.G.; Merfort, I.; Humar, M. Particulate Matter (PM2.5) from Biomass Combustion Induces an Anti-Oxidative Response and Cancer Drug Resistance in Human Bronchial Epithelial BEAS-2B Cells. Int. J. Environ. Res. Public Health 2020, 17, 8193. [Google Scholar] [CrossRef]
- An, J.; Wang, X.; Guo, P.; Zhong, Y.; Zhang, X.; Yu, Z. Hexabromocyclododecane and polychlorinated biphenyls increase resistance of hepatocellular carcinoma cells to cisplatin through the phosphatidylinositol 3-kinase/protein kinase B pathway. Toxicol. Lett. 2014, 229, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.A.; Valo, S.; Peltomäki, P.; Bajbouj, K.; Abdel-Rahman, W.M. Oncogenic Potential of Bisphenol A and Common Environmental Contaminants in Human Mammary Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 3735. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V.; Gómez-Guijosa, M.Á.; Cortes-Penagos, C. Acute Myeloid Leukemia-Genetic Alterations and Their Clinical Prognosis. Int. J. Hematol. Stem Cell Res. 2017, 11, 328–339. [Google Scholar]
- Lagunas-Rangel, F.A.; Bermúdez-Cruz, R.M. Natural Compounds That Target DNA Repair Pathways and Their Therapeutic Potential to Counteract Cancer Cells. Front. Oncol. 2020, 10, 598174. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Gnanasekar, M.; Rao, K.V.N.; Chen, L.; Narayanan, R.B.; Geetha, M.; Scott, A.L.; Ramaswamy, K.; Kaliraj, P. Molecular characterization of a calcium binding translationally controlled tumor protein homologue from the filarial parasites Brugia malayi and Wuchereria bancrofti. Mol. Biochem. Parasitol. 2002, 121, 107–118. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Agrawal, K. Vinblastine. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–4. [Google Scholar]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Ulukan, H.; Swaan, P.W. Camptothecins. Drugs 2002, 62, 2039–2057. [Google Scholar] [CrossRef]
- Pizzolato, J.F.; Saltz, L.B. The camptothecins. Lancet 2003, 361, 2235–2242. [Google Scholar] [CrossRef]
- Shagufta, W.; Ahmad, I. Tamoxifen a pioneering drug: An update on the therapeutic potential of tamoxifen derivatives. Eur. J. Med. Chem. 2018, 143, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.-H.; Chen, Z.-S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Cole, S.P.C.; Deeley, R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci. 2006, 27, 438–446. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Mungo, E.; Gazzano, E.; Kopecka, J.; Riganti, C. ERK is a Pivotal Player of Chemo-Immune-Resistance in Cancer. Int. J. Mol. Sci. 2019, 20, 2505. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Sethi, G. Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta-Rev. Cancer 2010, 1805, 167–180. [Google Scholar] [CrossRef]
- Capaccione, K.M.; Pine, S.R. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013, 34, 1420–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimple, R.C.; Wang, X. RAS: Striking at the Core of the Oncogenic Circuitry. Front. Oncol. 2019, 9, 965. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS Confers Chemoresistance by Upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakthivel, K.M.; Hariharan, S. Regulatory players of DNA damage repair mechanisms: Role in Cancer Chemoresistance. Biomed. Pharmacother. 2017, 93, 1238–1245. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A.; Bermúdez-Cruz, R.M. The Role of DNA Repair in Cellular Aging Process. In DNA Repair—An Update; IntechOpen: London, UK, 2019; ISBN 978-953-51-8070-8. [Google Scholar]
- Wong-Brown, M.W.; van der Westhuizen, A.; Bowden, N.A. Targeting DNA Repair in Ovarian Cancer Treatment Resistance. Clin. Oncol. 2020, 32, 518–526. [Google Scholar] [CrossRef]
- Alagpulinsa, D.A.; Ayyadevara, S.; Shmookler Reis, R.J. A Small-Molecule Inhibitor of RAD51 Reduces Homologous Recombination and Sensitizes Multiple Myeloma Cells to Doxorubicin. Front. Oncol. 2014, 4, 289. [Google Scholar] [CrossRef] [Green Version]
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/p53 Pathway Components Accelerates Tumor Development and Contributes to Radiation Resistance in Gliomas. Cancer Cell 2010, 18, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Guan, Y.; Chen, X.; Yang, J.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 2520. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O6-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2020, 9, 1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlang, B.; Falkner, K.C.; Cave, M.C.; Prough, R.A. Role of Cytochrome P450 Monooxygenase in Carcinogen and Chemotherapeutic Drug Metabolism. In Advances in Pharmacology; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 74, pp. 1–33. ISBN 9780128031193. [Google Scholar]
- Pathania, S.; Bhatia, R.; Baldi, A.; Singh, R.; Rawal, R.K. Drug metabolizing enzymes and their inhibitors’ role in cancer resistance. Biomed. Pharmacother. 2018, 105, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Ji, Y.; Jiang, X.; Qi, X. Downregulation of CYP2A6 and CYP2C8 in Tumor Tissues Is Linked to Worse Overall Survival and Recurrence-Free Survival from Hepatocellular Carcinoma. BioMed Res. Int. 2018, 2018, 5859415. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Steppi, A.; Zhou, Y.; Mao, F.; Miller, P.C.; He, M.M.; Zhao, T.; Sun, Q.; Zhang, J. Tumoral expression of drug and xenobiotic metabolizing enzymes in breast cancer patients of different ethnicities with implications to personalized medicine. Sci. Rep. 2017, 7, 4747. [Google Scholar] [CrossRef]
- Nunes, T.; Hamdan, D.; Leboeuf, C.; El Bouchtaoui, M.; Gapihan, G.; Nguyen, T.; Meles, S.; Angeli, E.; Ratajczak, P.; Lu, H.; et al. Targeting Cancer Stem Cells to Overcome Chemoresistance. Int. J. Mol. Sci. 2018, 19, 4036. [Google Scholar] [CrossRef] [Green Version]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V. FLT3–ITD and its current role in acute myeloid leukaemia. Med. Oncol. 2017, 34, 114. [Google Scholar] [CrossRef]
- Barbato, L.; Bocchetti, M.; Di Biase, A.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8, 926. [Google Scholar] [CrossRef] [Green Version]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Brown, Y.; Hua, S.; Tanwar, P.S. Extracellular matrix-mediated regulation of cancer stem cells and chemoresistance. Int. J. Biochem. Cell Biol. 2019, 109, 90–104. [Google Scholar] [CrossRef]
- Martins, S.G.; Zilhão, R.; Thorsteinsdóttir, S.; Carlos, A.R. Linking Oxidative Stress and DNA Damage to Changes in the Expression of Extracellular Matrix Components. Front. Genet. 2021, 12, 1279. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Fuentes-Retamal, S.; Palominos, C.; Rodríguez-Lucart, Y.A.; López-Torres, C.; Araya-Maturana, R. Extracellular Matrix Signals as Drivers of Mitochondrial Bioenergetics and Metabolic Plasticity of Cancer Cells During Metastasis. Front. Cell Dev. Biol. 2021, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Wazir, U.; Mokbel, K. Bisphenol A: A Concise Review of Literature and a Discussion of Health and Regulatory Implications. In Vivo 2019, 33, 1421–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seachrist, D.D.; Bonk, K.W.; Ho, S.-M.; Prins, G.S.; Soto, A.M.; Keri, R.A. A review of the carcinogenic potential of bisphenol A. Reprod. Toxicol. 2016, 59, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, S. Expression and DNA methylation changes in human breast epithelial cells after bisphenol A exposure. Int. J. Oncol. 2012, 41, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, S.; Keating, A.F. Bisphenol A-Induced Ovotoxicity Involves DNA Damage Induction to Which the Ovary Mounts a Protective Response Indicated by Increased Expression of Proteins Involved in DNA Repair and Xenobiotic Biotransformation. Toxicol. Sci. 2016, 152, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Finnish Society of Toxicology Abstracts of the 55th Congress of the European Societies of Toxicology (EUROTOX 2019) TOXICOLOGY SCIENCE PROVIDING SOLUTIONS. Toxicol. Lett. 2019, 314, S1–S309. [CrossRef]
- Matthews, J.B.; Twomey, K.; Zacharewski, T.R. In Vitro and in Vivo Interactions of Bisphenol A and Its Metabolite, Bisphenol A Glucuronide, with Estrogen Receptors α and β. Chem. Res. Toxicol. 2001, 14, 149–157. [Google Scholar] [CrossRef]
- George, O.; Bryant, B.K.; Chinnasamy, R.; Corona, C.; Arterburn, J.B.; Shuster, C.B. Bisphenol A Directly Targets Tubulin to Disrupt Spindle Organization in Embryonic and Somatic Cells. ACS Chem. Biol. 2008, 3, 167–179. [Google Scholar] [CrossRef]
- Alexandrov, K.; Rojas, M.; Satarug, S. The critical DNA damage by benzo(a)pyrene in lung tissues of smokers and approaches to preventing its formation. Toxicol. Lett. 2010, 198, 63–68. [Google Scholar] [CrossRef]
- Kasala, E.R.; Bodduluru, L.N.; Barua, C.C.; Sriram, C.S.; Gogoi, R. Benzo(a)pyrene induced lung cancer: Role of dietary phytochemicals in chemoprevention. Pharmacol. Rep. 2015, 67, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, R.; Gu, J.; Chen, Y.; Zhang, X.; Zhang, L.; Wu, H.; Hua, W.; Zeng, J. Aldehyde dehydrogenase 1A1 up-regulates stem cell markers in benzo[a]pyrene-induced malignant transformation of BEAS-2B cells. Environ. Toxicol. Pharmacol. 2016, 45, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, N.; Toyama, K.; Michihara, A.; Akasaki, K.; Tsuji, H.; Furuno, K. Effect of benzo[a]pyrene on P-glycoprotein-mediated transport in Caco-2 cell monolayer. Toxicology 2006, 223, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Pan, B.; Sakkiah, S.; Yavas, G.; Ge, W.; Zou, W.; Tong, W.; Hong, H. Persistent Organic Pollutants in Food: Contamination Sources, Health Effects and Detection Methods. Int. J. Environ. Res. Public Health 2019, 16, 4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alharbi, O.M.L.; Basheer, A.A.; Khattab, R.A.; Ali, I. Health and environmental effects of persistent organic pollutants. J. Mol. Liq. 2018, 263, 442–453. [Google Scholar] [CrossRef]
- Loury, D.J.; Byard, J.L. Aroclor 1254 pretreatment enhances the DNA repair response to amino acid pyrolysate mutagens in primary cultures of rat hepatocytes. Cancer Lett. 1983, 20, 283–290. [Google Scholar] [CrossRef]
- Shaddock, J.G.; Heflich, R.H.; McMillan, D.C.; Hinson, J.A.; Casciano, D.A. Pretreatment with mixed-function oxidase inducers increases the sensitivity of the hepatocyte/DNA repair assay. Environ. Mol. Mutagen. 1989, 13, 281–288. [Google Scholar] [CrossRef]
- Li, R.J.; Gao, H.; Na, G.S.; Lu, Z.H.; Yao, Y.; Yang, F. Hexabromocyclododecane-induced Genotoxicity in Cultured Human Breast Cells through DNA Damage. Biomed. Environ. Sci. 2017, 30, 296–300. [Google Scholar]
- Kim, S.-H.; Nam, K.-H.; Hwang, K.-A.; Choi, K.-C. Influence of hexabromocyclododecane and 4-nonylphenol on the regulation of cell growth, apoptosis and migration in prostatic cancer cells. Toxicol. Vitr. 2016, 32, 240–247. [Google Scholar] [CrossRef]
- Choi, K.-C. Cell growth of BG-1 ovarian cancer cells is promoted by di-n-butyl phthalate and hexabromocyclododecane via upregulation of the cyclin D and cyclin-dependent kinase-4 genes. Mol. Med. Rep. 2011, 5, 761–766. [Google Scholar] [CrossRef]
- Couleau, N.; Falla, J.; Beillerot, A.; Battaglia, E.; D’Innocenzo, M.; Plançon, S.; Laval-Gilly, P.; Bennasroune, A. Effects of Endocrine Disruptor Compounds, Alone or in Combination, on Human Macrophage-Like THP-1 Cell Response. PLoS ONE 2015, 10, e0131428. [Google Scholar] [CrossRef] [PubMed]
- Klotz, K.; Weistenhöfer, W.; Neff, F.; Hartwig, A.; van Thriel, C.; Drexler, H. The Health Effects of Aluminum Exposure. Dtsch. Aerzteblatt Online 2017, 114, 653–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondy, S.C. Low levels of aluminum can lead to behavioral and morphological changes associated with Alzheimer’s disease and age-related neurodegeneration. Neurotoxicology 2016, 52, 222–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farasani, A.; Darbre, P.D. Effects of aluminium chloride and aluminium chlorohydrate on DNA repair in MCF10A immortalised non-transformed human breast epithelial cells. J. Inorg. Biochem. 2015, 152, 186–189. [Google Scholar] [CrossRef]
- Mandriota, S.J.; Tenan, M.; Ferrari, P.; Sappino, A. Aluminium chloride promotes tumorigenesis and metastasis in normal murine mammary gland epithelial cells. Int. J. Cancer 2016, 139, 2781–2790. [Google Scholar] [CrossRef]
- Saraç, N.; Uğur, A.; Karaca, İ.R. Evaluation of antioxidant and antimutagenic activities of aluminum chloride. Eur. Oral Res. 2019, 53, 51–55. [Google Scholar] [CrossRef]
- Kim, K.-H.; Kabir, E.; Kabir, S. A review on the human health impact of airborne particulate matter. Environ. Int. 2015, 74, 136–143. [Google Scholar] [CrossRef]
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and Health Impacts of Air Pollution: A Review. Front. Public Health 2020, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhong, Y.; Hou, T.; Liao, J.; Zhang, C.; Sun, C.; Wang, G. PM2.5 induces EMT and promotes CSC properties by activating Notch pathway in vivo and vitro. Ecotoxicol. Environ. Saf. 2019, 178, 159–167. [Google Scholar] [CrossRef]
- Wei, H.; Liang, F.; Cheng, W.; Zhou, R.; Wu, X.; Feng, Y.; Wang, Y. The mechanisms for lung cancer risk of PM 2.5: Induction of epithelial-mesenchymal transition and cancer stem cell properties in human non-small cell lung cancer cells. Environ. Toxicol. 2017, 32, 2341–2351. [Google Scholar] [CrossRef]
- Heßelbach, K.; Kim, G.-J.; Flemming, S.; Häupl, T.; Bonin, M.; Dornhof, R.; Günther, S.; Merfort, I.; Humar, M. Disease relevant modifications of the methylome and transcriptome by particulate matter (PM 2.5) from biomass combustion. Epigenetics 2017, 12, 779–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Pollutant | DN(M)EL Long-Term Exposure | Concentration Tested | Chemotherapeutic Drug | Cancer | Model | Mechanism Associated with Decreased Efficacy of the Chemotherapeutic Drug | Reference | ||
|---|---|---|---|---|---|---|---|---|---|
| Inhalation a | Dermal b | Oral b | |||||||
| BPA | --- | 0.042 μg c | 4 μg c | 1 nM/48 h | CDDP ≤400 ng/mL/24 h | BC | T47D cells MDA-MB-468 cells |
| [12,13] |
| 1 nM/48 h | DOX ≤125 ng/mL/24 h | BC | T47D cells MDA-MB-468 cells |
| [12] | ||||
| 1 nM/48 h | VIN 1 ng/mL/24 h | BC | T47D cells MDA-MB-468 cells | --- | [12] | ||||
| 4 μM/48 h | DOX 4.4 μM/24 h | COAD | HT29 cells |
| [14] | ||||
| ≤4.4 µM/48 h | DOX 4 μM/24 h | --- | MRC-5 cells |
| [15] | ||||
| 150 μM/24 h | CPT 100 nM/24 h | --- | MEF cells |
| [16] | ||||
| BaP | 1.43 µg c | --- | 0.5 µg c | 10 μM/24 h | CDDP 4.2 μM/24 h | OC | WHCO1 cells WHCO5 cells |
| [17] |
| 10 μM/24 h | 5-FU 3.5 μM/24 h | OC | WHCO1 cells WHCO5 cells |
| [17] | ||||
| 10 μM/24 h | PTX 2 μM/24 h | OC | WHCO1 cells WHCO5 cells |
| [17] | ||||
| 10 μM/24 h | CDDP + 5-FU 4.2 μM + 3.5 μM/24 h | OC | WHCO1 cells |
| [17] | ||||
| 10 μM/24 h | CDDP + PTX 4.2 μM + 2 μM/24 h | OC | WHCO1 cells |
| [17] | ||||
| 10 μM/24 h | 5-FU + PTX 3.5 μM + 2 μM/24 h | OC | WHCO1 cells |
| [17] | ||||
| 50 nM/3 months | CDDP ≤100 μM/48 h | SCC | CAL27 cells SCC9 cells | --- | [18] | ||||
| 50 nM/3 months | 5-FU ≤100 μg/mL/48 h | SCC | CAL27 cells SCC9 cells | --- | [18] | ||||
| AlCl3 | 4 mg c | 2.32 mg c | 2.3 mg c | ≤200 μM/96 h | 5-FU 100 μM/48 h | HCC | HepG2 cells |
| [19] |
| PM2.5 | 25 μg d | --- | --- | 100 μg/mL/5 weeks | DOX 1 μM/48 h | --- | BEAS-2B cells |
| [20] |
| HBCD | 719 μg c | 1020 mg c | 102 μg c | 1 μM/48 h | CDDP 10 μM/24 h | HCC | HepG2 cells MHCC97H cells |
| [21] |
| 0.0015 nM/2 months | DOX CNM/12 h | --- | HME1 cells | --- | [22] | ||||
| PCB-1254 | --- | --- | --- | 1 μg/mL/48 h | CDDP 10 μM/24 h | HCC | HepG2 cells MHCC97H cells |
| [21] |
| OP | 0.6 mg c | 5.6 mg c | 0.1 mg c | 0.0048 nM/2 months | DOX CNM/12 h | --- | HME1 cells | --- | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagunas-Rangel, F.A.; Liu, W.; Schiöth, H.B. Can Exposure to Environmental Pollutants Be Associated with Less Effective Chemotherapy in Cancer Patients? Int. J. Environ. Res. Public Health 2022, 19, 2064. https://doi.org/10.3390/ijerph19042064
Lagunas-Rangel FA, Liu W, Schiöth HB. Can Exposure to Environmental Pollutants Be Associated with Less Effective Chemotherapy in Cancer Patients? International Journal of Environmental Research and Public Health. 2022; 19(4):2064. https://doi.org/10.3390/ijerph19042064
Chicago/Turabian StyleLagunas-Rangel, Francisco Alejandro, Wen Liu, and Helgi B. Schiöth. 2022. "Can Exposure to Environmental Pollutants Be Associated with Less Effective Chemotherapy in Cancer Patients?" International Journal of Environmental Research and Public Health 19, no. 4: 2064. https://doi.org/10.3390/ijerph19042064