
Bacterial Community Composition and Function of Tropical River Ecosystem along the Nandu River on Hainan Island, China
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Water Property
2.3. DNA Extraction, PCR Amplification, and Sequencing
2.4. Sequenced Data Processing
2.5. Statistical Analyses
3. Results
3.1. Properties of Water Samples
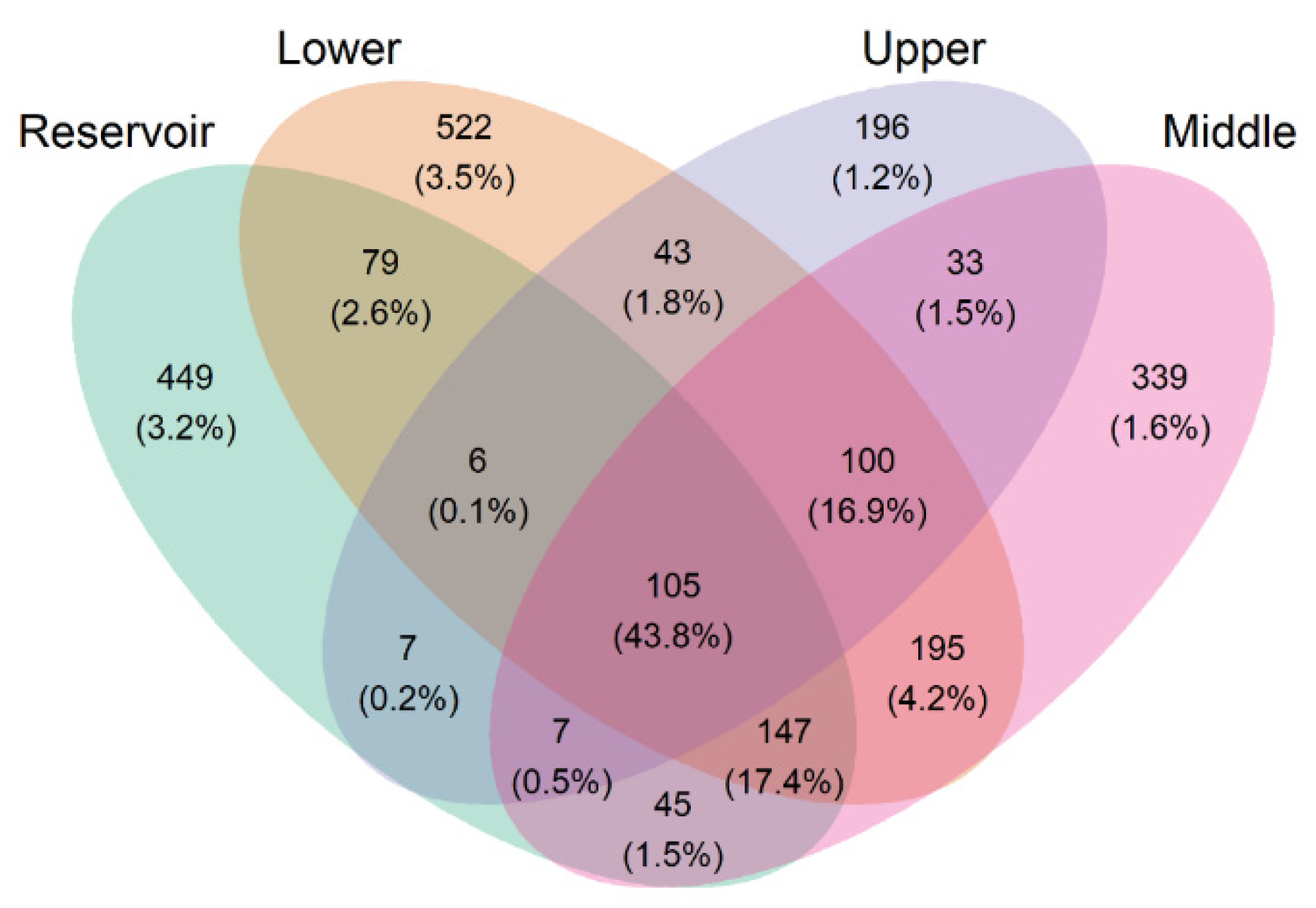
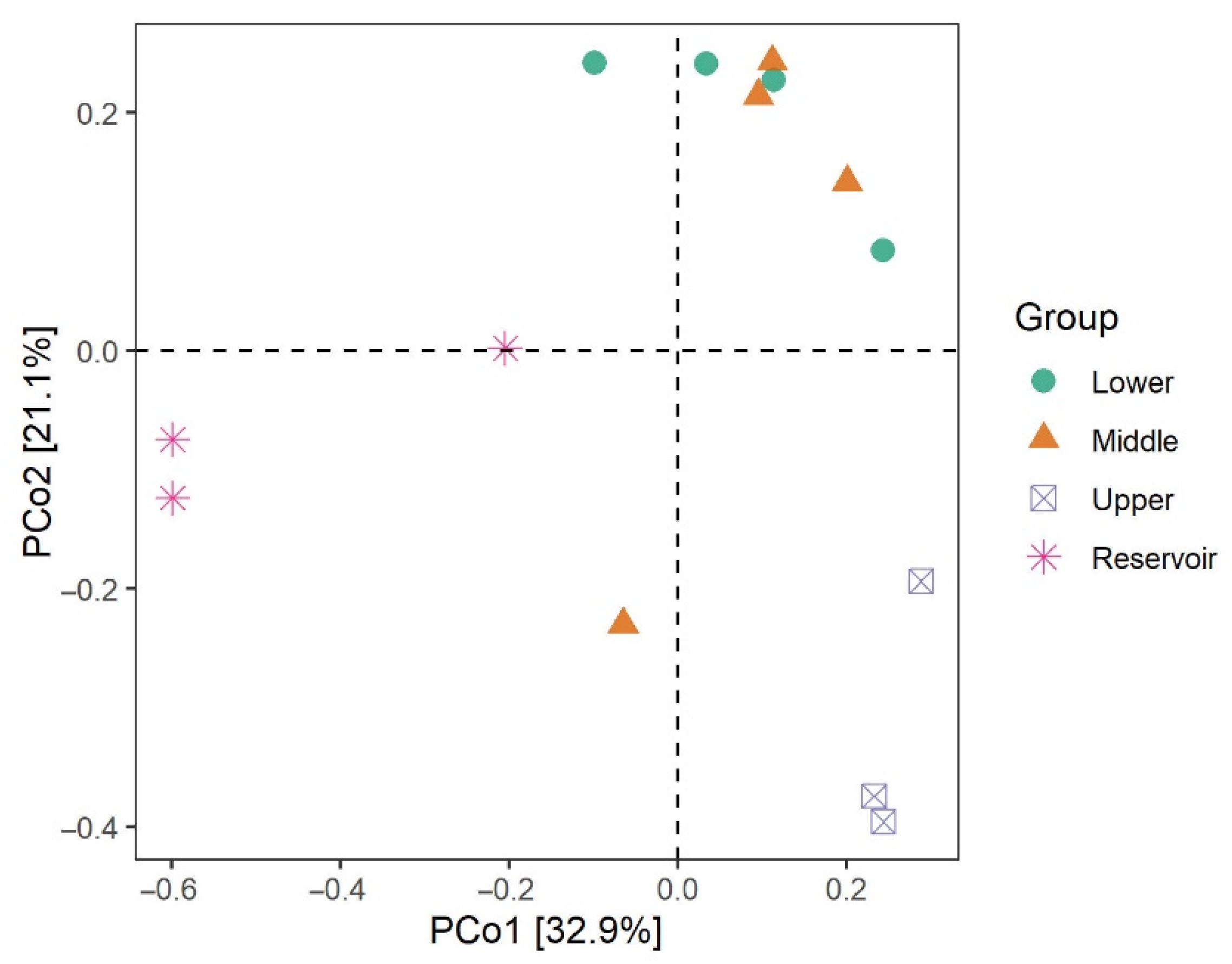
3.2. The Bacterial Diversity of the Surface Water
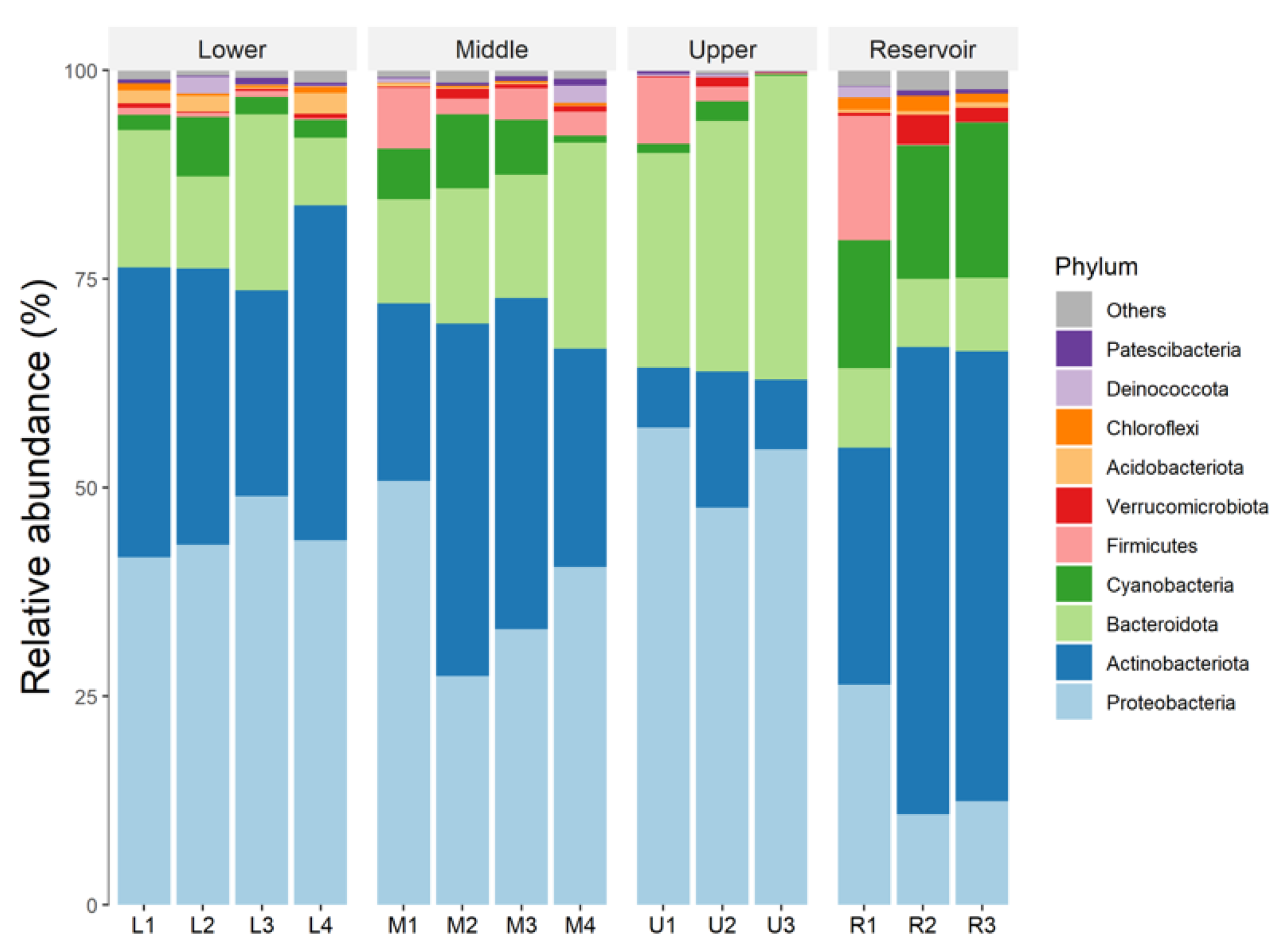
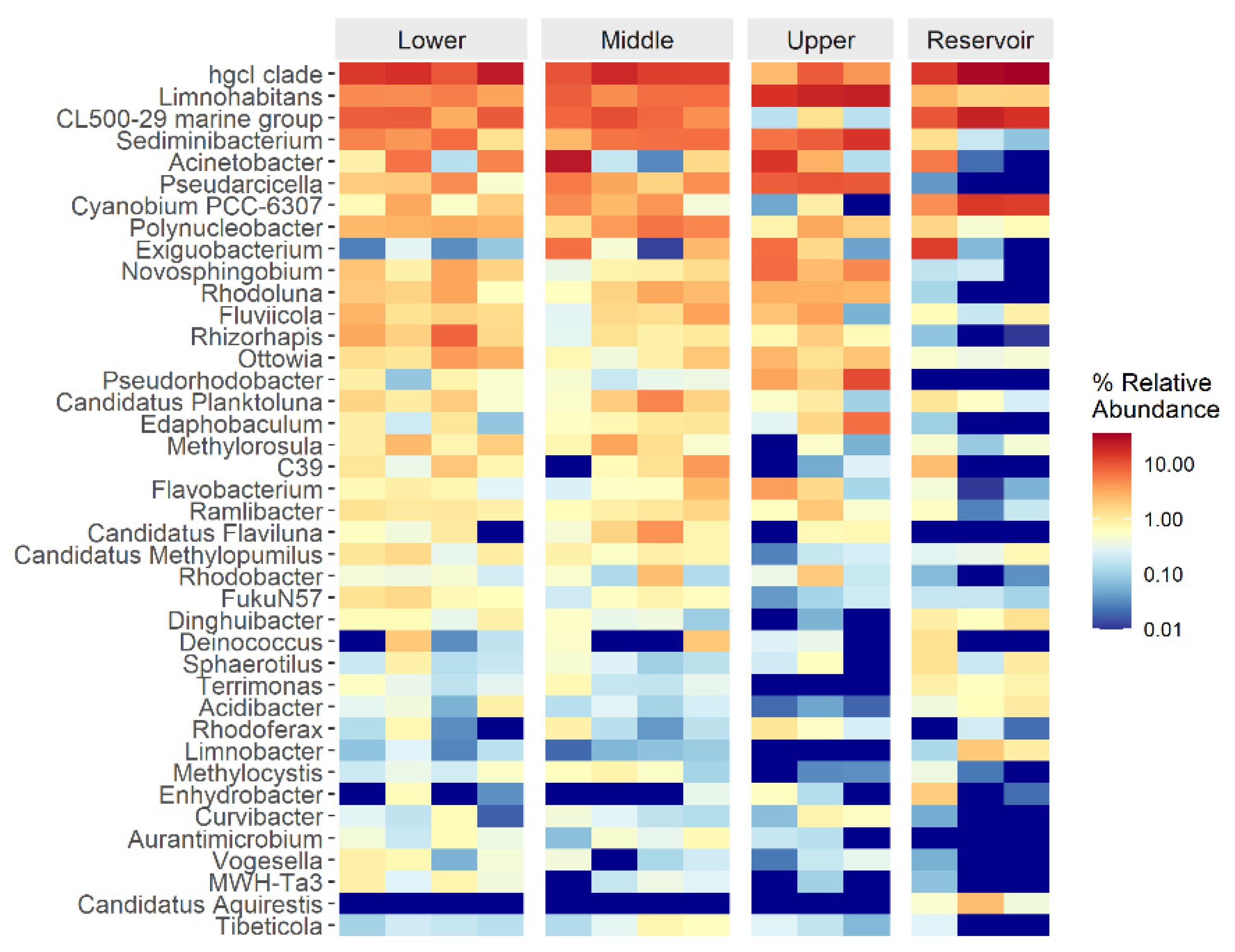
3.3. Bacterial Community Structure and Composition in the Water Samples
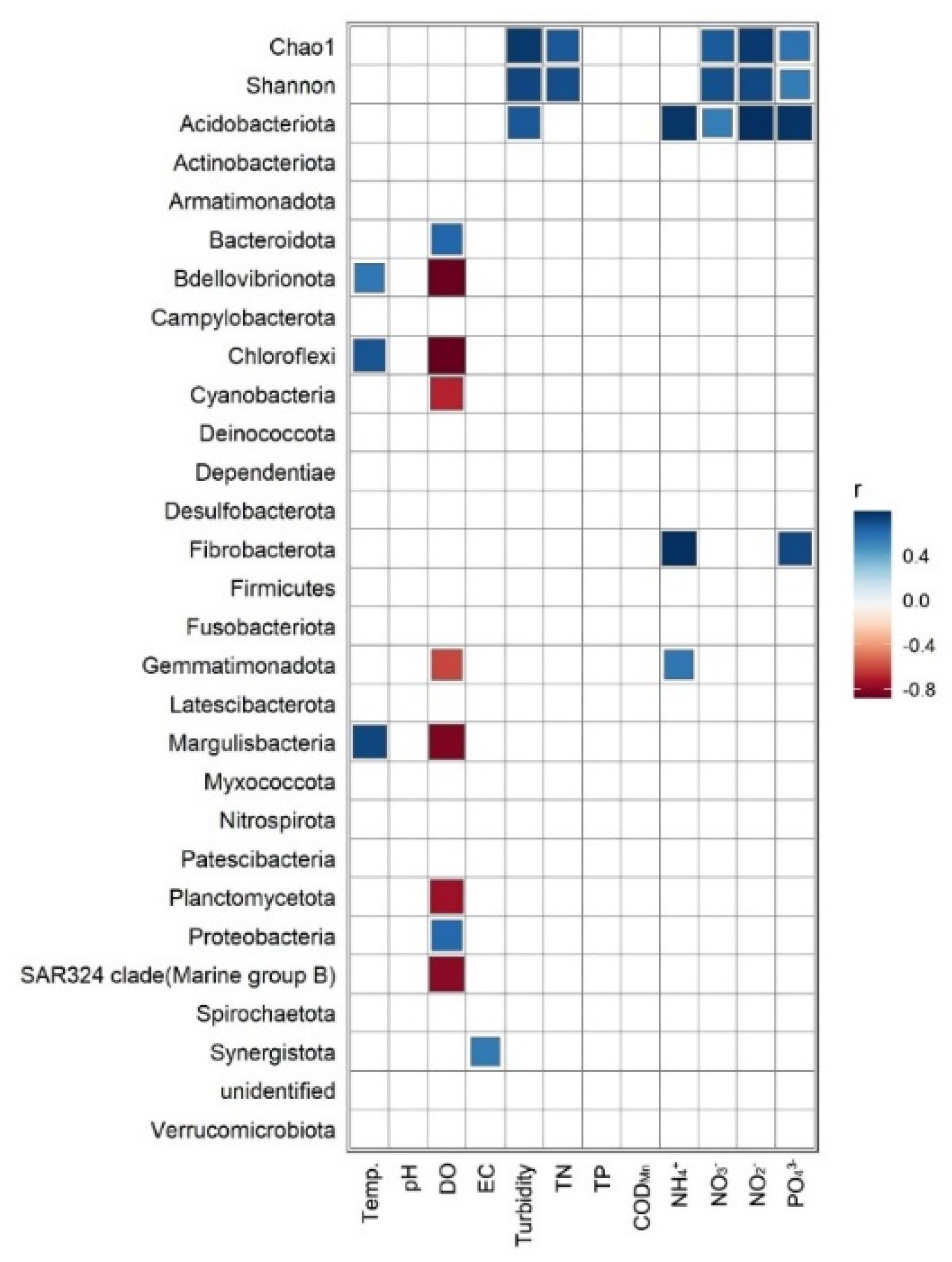
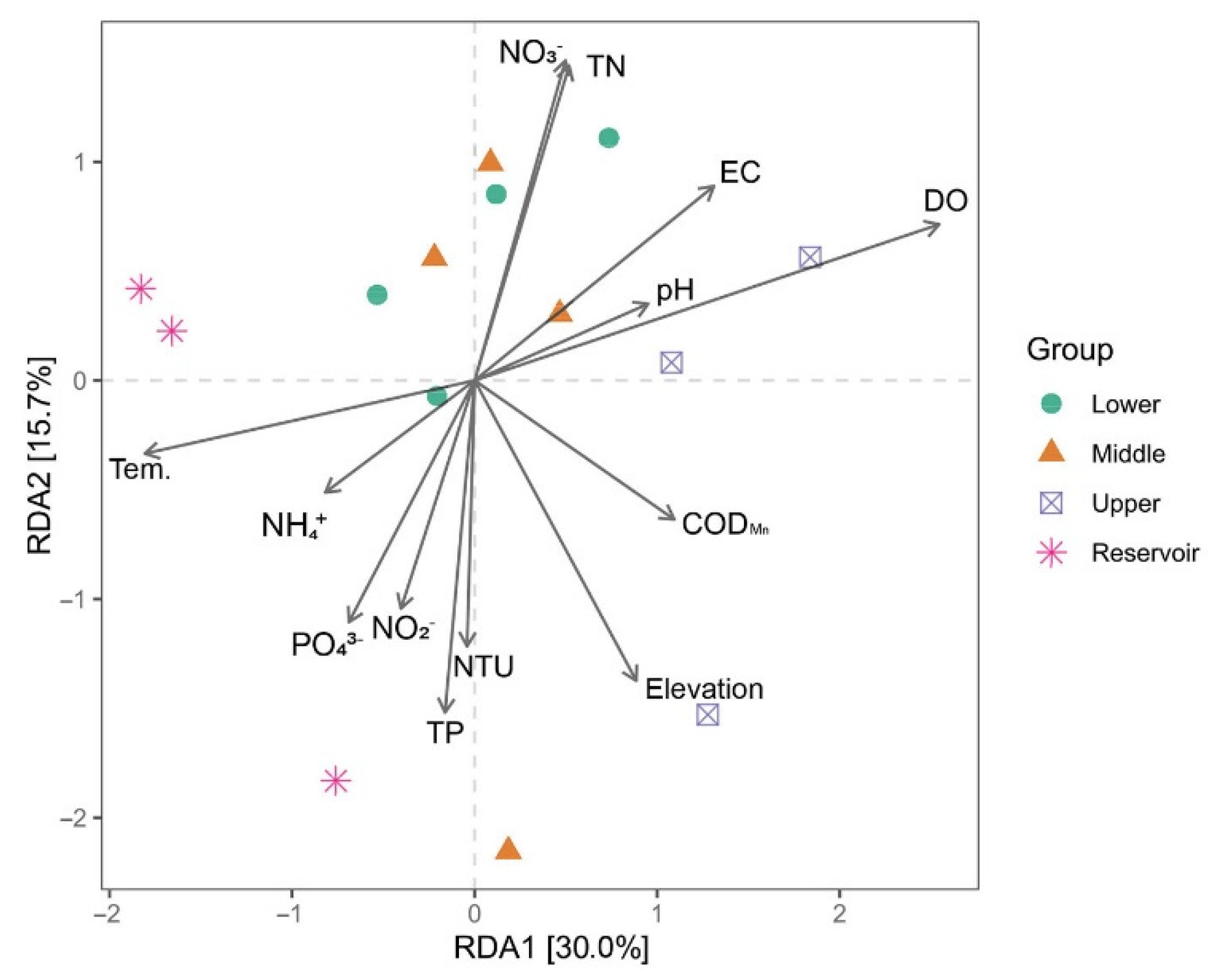
3.4. Correlation between Bacterial Communities and Environmental Factors
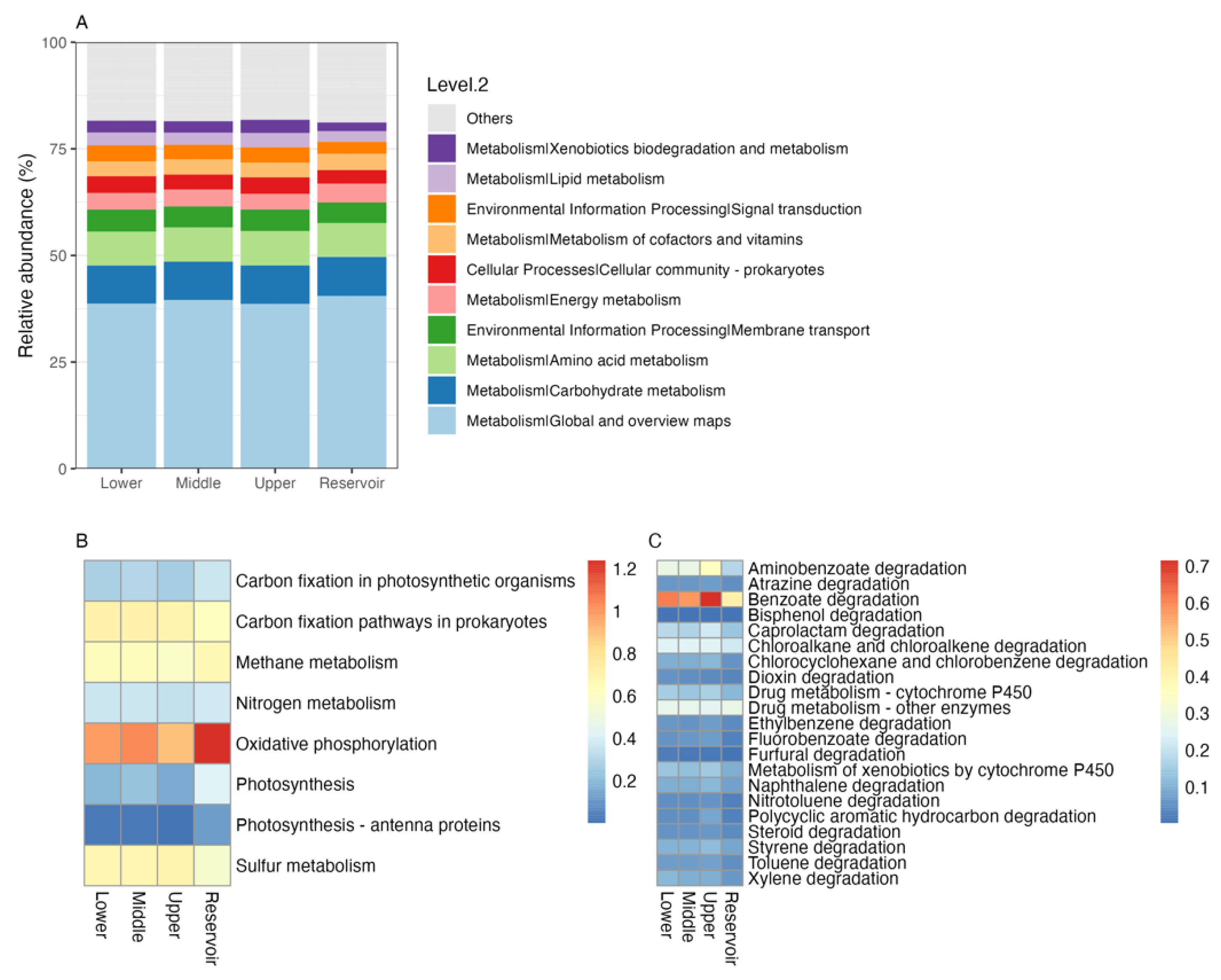
3.5. Bacterial Functional Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Giller, P.S.; Giller, P.; Malmqvist, B.; Department of Animal Ecology Bj Rn Malmqvist. The Biology of Streams and Rivers; OUP Oxford: Oxford, UK, 1998. [Google Scholar]
- Cooper, C.M. Biological Effects of Agriculturally Derived Surface Water Pollutants on Aquatic Systems—A Review. J. Environ. Qual. 1993, 22, 402–408. [Google Scholar] [CrossRef]
- Okereafor, U.; Makhatha, M.; Mekuto, L.; Uche-Okereafor, N.; Sebola, T.; Mavumengwana, V. Toxic Metal Implications on Agricultural Soils, Plants, Animals, Aquatic Life and Human Health. Int. J. Environ. Res. Public Health 2020, 17, 2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashmi, I.; Roy, T.; Kartika, K.S.; Pal, R.; Coumar, V.; Kala, S.; Shinoji, K.C. Organic and Inorganic Fertilizer Contaminants in Agriculture: Impact on Soil and Water Resources. In Contaminants in Agriculture; Springer: Berlin/Heidelberg, Germany, 2020; pp. 3–41. [Google Scholar]
- Cabral, J.P.S. Water Microbiology. Bacterial Pathogens and Water. Int. J. Environ. Res. Public Health 2010, 7, 3657–3703. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Diamond, J. China’s Environment in a Globalizing World. Nature 2005, 435, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guidelines for Drinking-Water Quality; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Zeglin, L.H. Stream Microbial Diversity in Response to Environmental Changes: Review and Synthesis of Existing Research. Front. Microbiol. 2015, 6, 454. [Google Scholar] [CrossRef] [Green Version]
- Department of Hainan Province Ocean and Fishery. Hainan Province Marine Environment Condition Bulletin in 2016. 2017. Available online: http://cn.chinagate.cn/environment/2018-03/17/content_50684722.htm (accessed on 10 August 2022).
- Zhang, J.; Zhang, P.; Dai, P.; Lai, J.; Chen, Y. Spatiotemporal Distributions of DIP and the Eutrophication in Hainan Island Adjacent Coastal Water. China Environ. Sci. 2019, 39, 2541–2548. (In Chinese) [Google Scholar]
- Zhang, P.; Wei, L.; Lai, J.; Dai, P.; Chen, Y.; Zhang, J. Concentration, Composition and Fluxes of Land-Based Nitrogen and Phosphorus Source Pollutants Input into Zhanjiang Bay in Summer. J. Guangdong Ocean Univ. 2019, 39, 46–55. (In Chinese) [Google Scholar]
- Zhao, D.; Wan, S.; Yu, Z.; Huang, J. Distribution, Enrichment and Sources of Heavy Metals in Surface Sediments of Hainan Island Rivers, China. Environ. Earth Sci. 2015, 74, 5097–5110. [Google Scholar] [CrossRef]
- Saxena, G.; Bharagava, R.N.; Kaithwas, G.; Raj, A. Microbial Indicators, Pathogens and Methods for Their Monitoring in Water Environment. J. Water Health 2014, 13, 319–339. [Google Scholar] [CrossRef] [Green Version]
- Toze, S. PCR and the Detection of Microbial Pathogens in Water and Wastewater. Water Res. 1999, 33, 3545–3556. [Google Scholar] [CrossRef]
- Semedo-Aguiar, A.P.; Pereira-Leal, J.B.; Leite, R.B. Microbial Diversity and Toxin Risk in Tropical Freshwater Reservoirs of Cape Verde. Toxins 2018, 10, 186. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.; Ng, C.; Nshimyimana, J.P.; Loh, L.L.; Gin, K.Y.-H.; Thompson, J.R. Next-Generation Sequencing (NGS) for Assessment of Microbial Water Quality: Current Progress, Challenges, and Future Opportunities. Front. Microbiol. 2015, 6, 1027. [Google Scholar] [CrossRef]
- Aguilar, P.; Dorador, C.; Vila, I.; Sommaruga, R. Bacterioplankton Composition in Tropical High-Elevation Lakes of the Andean Plateau. FEMS Microbiol. Ecol. 2018, 94, fiy004. [Google Scholar] [CrossRef] [Green Version]
- Godoy, R.G.; Marcondes, M.A.; Pessôa, R.; Nascimento, A.; Victor, J.R.; Duarte, A.J.d.S.; Clissa, P.B.; Sanabani, S.S. Bacterial Community Composition and Potential Pathogens along the Pinheiros River in the Southeast of Brazil. Sci. Rep. 2020, 10, 9331. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Li, H.; Yang, H.; Peng, C.; Peng, Z.; Lu, L. Shift in the Microbial Community Composition of Surface Water and Sediment along an Urban River. Sci. Total Environ. 2018, 627, 600–612. [Google Scholar] [CrossRef]
- Gong, J.; Gao, J.; Fu, Y.; Tang, S.; Cai, Y.; Yang, J.; Wu, H.; Ma, S. Vertical Distribution and Major Influencing Factors of Soil Selenium in Tropical Climate: A Case Study of Chengmai County, Hainan Island. Chemosphere 2023, 312, 137207. [Google Scholar] [CrossRef]
- Li, J.; Li, N.; Li, F.; Zou, T.; Yu, S.; Wang, Y.; Qin, S.; Wang, G. Spatial Diversity of Bacterioplankton Communities in Surface Water of Northern South China Sea. PLoS ONE 2014, 9, e113014. [Google Scholar] [CrossRef]
- Chen, D.; Ding, J. Study on Influencing Factors of Measurement of Total Nitrogen by Digestion with UV-Alkaline Potassium Persulfate and Reduction with Hydrazine Sulphate Spectrophotometric Method and Application. In Proceedings of the 2008 2nd International Conference on Bioinformatics and Biomedical Engineering, Shanghai, China, 16–18 May 2008; pp. 3223–3227. [Google Scholar]
- Yang, C.-X.; Sun, X.-Y.; Liu, B.; Lian, H.-T. Determination of Total Phosphorus in Water Sample by Digital Imaging Colorimetry. Chin. J. Anal. Chem. 2007, 35, 850–853. [Google Scholar] [CrossRef]
- International Organization for Standardization. Water Quality—Determination of Permanganate Index; International Organization for Standardization: Geneva, Switzerland, 1993. [Google Scholar]
- Feng, Y.; Zhao, Y.; Guo, Y.; Liu, S. Microbial Transcript and Metabolome Analysis Uncover Discrepant Metabolic Pathways in Autotrophic and Mixotrophic Anammox Consortia. Water Res. 2018, 128, 402–411. [Google Scholar] [CrossRef]
- Callahan, B.J.; Sankaran, K.; Fukuyama, J.A.; McMurdie, P.J.; Holmes, S.P. Bioconductor Workflow for Microbiome Data Analysis: From Raw Reads to Community Analyses. F1000Res 2016, 5, 1492. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of Habitat-Specific Functional Profiles and Functional Redundancy Based on 16S rRNA Gene Sequences. Environ. Microbiome 2020, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J. Vegan: Ecological Diversity. R Project 2018. Available online: https://cran.r-project.org/web/packages/vegan/gnettes/diversity-vegan.pdf (accessed on 20 April 2022).
- Abdi, H.; Williams, L.J. Principal Component Analysis: Principal Component Analysis. WIREs Comp. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; American Cancer Society: Atlanta, GA, USA, 2017; pp. 1–15. [Google Scholar]
- R Core Team. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2018. [Google Scholar]
- Jin, D.; Kong, X.; Cui, B.; Jin, S.; Xie, Y.; Wang, X.; Deng, Y. Bacterial Communities and Potential Waterborne Pathogens within the Typical Urban Surface Waters. Sci. Rep. 2018, 8, 13368. [Google Scholar] [CrossRef] [Green Version]
- Betiku, O.C.; Sarjeant, K.C.; Ngatia, L.W.; Aghimien, M.O.; Odewumi, C.O.; Latinwo, L.M. Evaluation of Microbial Diversity of Three Recreational Water Bodies Using 16S rRNA Metagenomic Approach. Sci. Total Environ. 2021, 771, 144773. [Google Scholar] [CrossRef]
- Burkert, U.; Warnecke, F.; Babenzien, D.; Zwirnmann, E.; Pernthaler, J. Members of a Readily Enriched β-Proteobacterial Clade Are Common in Surface Waters of a Humic Lake. Appl. Environ. Microbiol. 2003, 69, 6550–6559. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.J.; Jones, S.E.; Eiler, A.; McMahon, K.D.; Bertilsson, S. A Guide to the Natural History of Freshwater Lake Bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ding, X.; Guan, R.; Zhu, C.; Xu, C.; Zhu, B.; Zhang, H.; Xiong, Z.; Xue, Y.; Tu, J. Evaluation of Different 16S rRNA Gene V Regions for Exploring Bacterial Diversity in a Eutrophic Freshwater Lake. Sci. Total Environ. 2018, 618, 1254–1267. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, M.; Huang, T.; Miao, Y.; Li, H.; Liu, K.; Yang, W.; Ma, B. Spatial and Temporal Dynamics of Actinobacteria in Drinking Water Reservoirs: Novel Insights into Abundance, Community Structure, and Co-Existence Model. Sci. Total Environ. 2022, 814, 152804. [Google Scholar] [CrossRef]
- Liu, J.; Fu, B.; Yang, H.; Zhao, M.; He, B.; Zhang, X.-H. Phylogenetic Shifts of Bacterioplankton Community Composition along the Pearl Estuary: The Potential Impact of Hypoxia and Nutrients. Front. Microbiol. 2015, 6, 64. [Google Scholar] [CrossRef]
- Warnecke, F.; Amann, R.; Pernthaler, J. Actinobacterial 16S rRNA Genes from Freshwater Habitats Cluster in Four Distinct Lineages. Environ. Microbiol. 2004, 6, 242–253. [Google Scholar] [CrossRef]
- Warnecke, F.; Sommaruga, R.; Sekar, R.; Hofer, J.S.; Pernthaler, J. Abundances, Identity, and Growth State of Actinobacteria in Mountain Lakes of Different UV Transparency. Appl. Environ. Microbiol. 2005, 71, 5551–5559. [Google Scholar] [CrossRef] [Green Version]
- Domingues, R.B.; Barbosa, A.B.; Sommer, U.; Galvão, H.M. Phytoplankton Composition, Growth and Production in the Guadiana Estuary (SW Iberia): Unraveling Changes Induced after Dam Construction. Sci. Total Environ. 2012, 416, 300–313. [Google Scholar] [CrossRef]
- Gao, G.; Xiao, L.; Lin, Q.; Hu, R.; Lei, L. Structure of phytoplankton functional groups and water quality assessment of main reservoirs in Hainan Province. Ecol. Sci. 2013, 32, 144–150. (In Chinese) [Google Scholar]
- Wang, S.; Hou, W.; Jiang, H.; Huang, L.; Dong, H.; Chen, S.; Wang, B.; Chen, Y.; Lin, B.; Deng, Y. Microbial Diversity Accumulates in a Downstream Direction in the Three Gorges Reservoir. J. Environ. Sci. 2021, 101, 156–167. [Google Scholar] [CrossRef]
- Pikaar, I.; Matassa, S.; Rabaey, K.; Bodirsky, B.L.; Popp, A.; Herrero, M.; Verstraete, W. Microbes and the Next Nitrogen Revolution. Environ. Sci. Technol. 2017, 51, 7297–7303. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.K. Water Quality: Science, Assessments and Policy; Open access peer-reviewed edited volume, IntechOpen Book Series; U.S. Environmental Protection Agency: Washington, DC, USA, 2020. [Google Scholar]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: A R-Based Tool for the Rapid Prediction of Habitat-Specific Functional Profiles and Functional Redundancy Based on 16S rRNA Gene Marker Gene Sequences. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Li, Y.; Zhang, W.; Wang, C.; Wang, P.; Niu, L.; Du, J.; Gao, Y. Bacterial Community Composition and Function Shift with the Aggravation of Water Quality in a Heavily Polluted River. J. Environ. Manag. 2019, 237, 433–441. [Google Scholar] [CrossRef]
- Dindhoria, K.; Kumar, S.; Kumar, R. Taxonomic and Functional Analysis of Proglacial Water Bodies of Triloknath Glacier Ecosystem from North-Western Himalayas. Ecol. Inform. 2021, 64, 101365. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Reservoir | Upper | Middle | Lower |
|---|---|---|---|---|
| Temperature (°C) | 25.87 ± 1.26 a | 23.57 ± 0.60 ab | 23.33 ± 1.19 b | 25.00 ± 0.91 ab |
| pH | 7.20 ± 0.30 a | 7.50 ± 0.48 a | 7.29 ± 0.15 a | 7.35 ± 0.29 a |
| DO (mg L−1) | 7.05 ± 0.76 b | 8.53 ± 0.16 a | 8.47 ± 0.10 a | 8.08 ± 0.28 a |
| EC (uS cm−1) | 81.27 ± 16.31 ab | 96.37 ± 11.63 ab | 78.30 ± 19.68 b | 117.28 ± 11.90 a |
| Turbidity | 4.36 ± 2.00 b | 4.24 ± 1.19 b | 11.33 ± 2.95 ab | 17.13 ± 4.85 a |
| TN (mg L−1) | 0.86 ± 0.20 c | 1.84 ± 1.06 bc | 4.75 ± 2.00 ab | 6.28 ± 1.68 a |
| TP (mg L−1) | 0.10 ± 0.05 a | 0.13 ± 0.06 a | 0.15 ± 0.06 a | 0.17 ± 0.07 a |
| CODMn (mg L−1) | 0.75 ± 0.18 a | 4.69 ± 5.91 a | 1.28 ± 0.91 a | 2.88 ± 3.68 a |
| NH4+ (mg L−1) | 0.05 ± 0.03 a | 0.04 ± 0.04 a | 0.06 ± 0.03 a | 0.29 ± 0.34 a |
| NO3− (mg L−1) | 0.59 ± 0.49 c | 1.56 ± 1.31 bc | 4.43 ± 2.04 ab | 6.07 ± 1.52 a |
| NO2− (mg L−1) | 0.02 ± 0.03 b | 0.01 ± 0.01 b | 0.03 ± 0.02 b | 0.09 ± 0.02 a |
| PO43− (mg L−1) | 0.03 ± 0.01 a | 0.02 ± 0.01 a | 0.08 ± 0.05 a | 0.14 ± 0.09 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhai, Y.; Ge, G.; Xu, Y.; Wang, C.; Hu, A.; Han, Y.; Shan, N.; Liu, B.; Chen, J.; et al. Bacterial Community Composition and Function of Tropical River Ecosystem along the Nandu River on Hainan Island, China. Int. J. Environ. Res. Public Health 2023, 20, 382. https://doi.org/10.3390/ijerph20010382
Li J, Zhai Y, Ge G, Xu Y, Wang C, Hu A, Han Y, Shan N, Liu B, Chen J, et al. Bacterial Community Composition and Function of Tropical River Ecosystem along the Nandu River on Hainan Island, China. International Journal of Environmental Research and Public Health. 2023; 20(1):382. https://doi.org/10.3390/ijerph20010382
Chicago/Turabian StyleLi, Jinbiao, Yangni Zhai, Guojian Ge, Yang Xu, Can Wang, Anyong Hu, Yujie Han, Nan Shan, Bo Liu, Jinlin Chen, and et al. 2023. "Bacterial Community Composition and Function of Tropical River Ecosystem along the Nandu River on Hainan Island, China" International Journal of Environmental Research and Public Health 20, no. 1: 382. https://doi.org/10.3390/ijerph20010382