
Ecogeographic Drivers of the Spatial Spread of Highly Pathogenic Avian Influenza Outbreaks in Europe and the United States, 2016–Early 2022
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Time-Scaled Bayesian Phylogenetic Analyses
2.3. Discrete Trait Diffusion Analyses between Geographic Regions
2.4. Generalized Linear Models and Empirical Predictors
3. Results
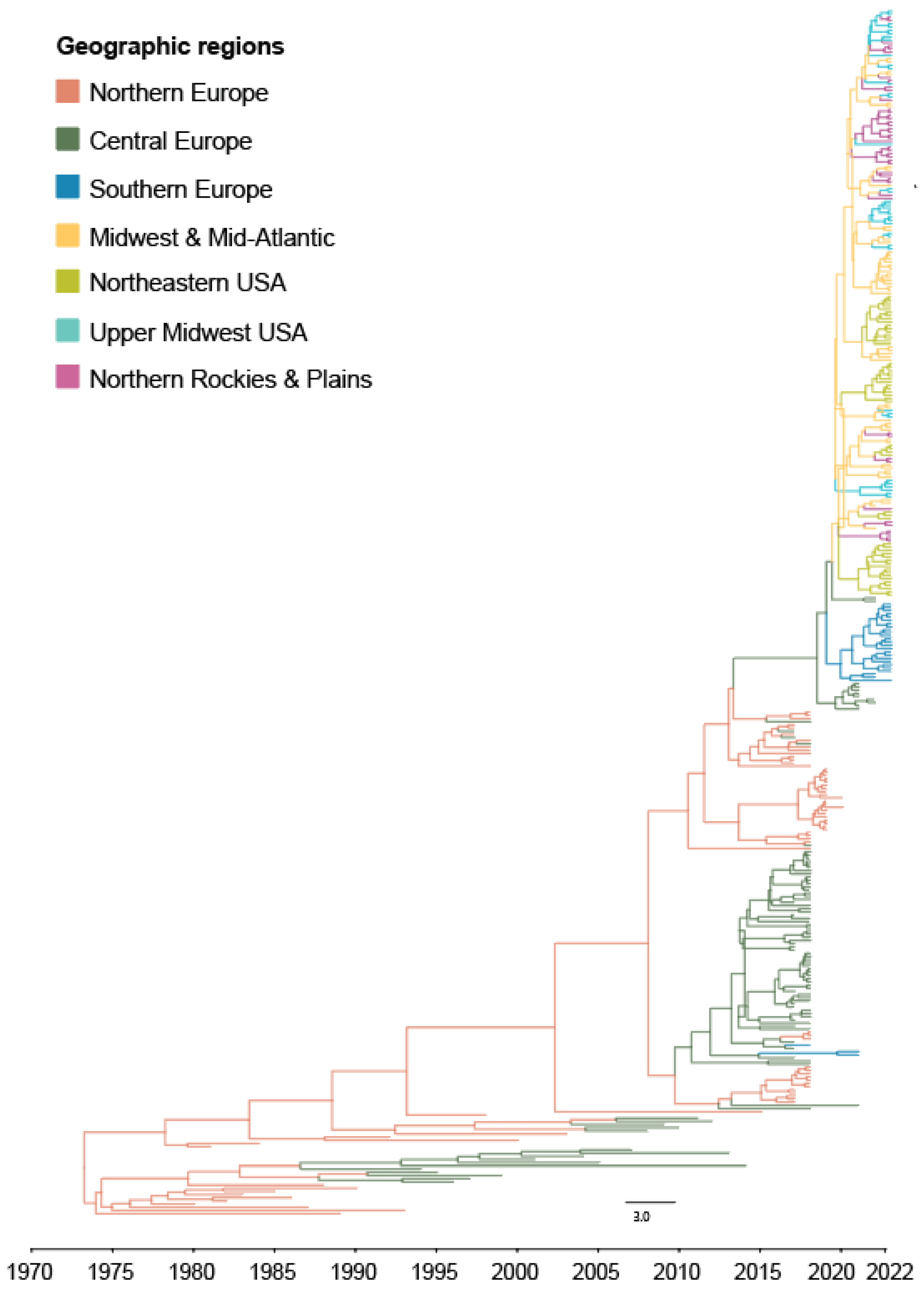
3.1. Bayesian Phylogeography of the H5Nx HPAI Virus Clade 2.3.4.4 Outbreak in Europe and United States, 2016 to Early 2022
3.2. Generalized Linear Model of Ecological and Environmental Predictors of H5Nx Diffusion in Europe and the US, 2016–2022
4. Discussion
4.1. Limitations
4.2. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, D.-H.; Bertran, K.; Kwon, J.-H.; Swayne, D.E. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J. Vet. Sci. 2017, 18, 269–280. [Google Scholar] [CrossRef]
- Antigua, K.J.C.; Choi, W.-S.; Baek, Y.H.; Song, M.-S. The Emergence and Decennary Distribution of Clade 2.3.4.4 HPAI H5Nx. Microorganisms 2019, 7, 156. [Google Scholar] [CrossRef]
- Dhingra, M.S.; Artois, J.; Robinson, T.P.; Linard, C.; Chaiban, C.; Xenarios, I.; Engler, R.; Liechti, R.; Kuznetsov, D.; Xiao, X.; et al. Global mapping of highly pathogenic avian influenza H5N1 and H5Nx clade 2.3.4.4 viruses with spatial cross-validation. eLife 2016, 5, e19571. [Google Scholar] [CrossRef]
- Hill, N.J.; Hussein, I.T.; Davis, K.R.; Ma, E.J.; Spivey, T.J.; Ramey, A.M.; Puryear, W.B.; Das, S.R.; Halpin, R.A.; Lin, X.; et al. Reassortment of Influenza A Viruses in Wild Birds in Alaska before H5 Clade 2.3.4.4 Outbreaks. Emerg. Infect. Dis. 2017, 23, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Torchetti, M.K.; Killian, M.L.; Deliberto, T.J.; Swayne, D. Reoccurrence of Avian Influenza A(H5N2) Virus Clade 2.3.4.4 in Wild Birds, Alaska, USA, 2016. Emerg. Infect. Dis. 2017, 23, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Sharshov, K.; Swayne, D.; Kurskaya, O.; Sobolev, I.; Kabilov, M.; Alekseev, A.; Irza, V.; Shestopalov, A. Novel Reassortant Clade 2.3.4.4 Avian Influenza A(H5N8) Virus in Wild Aquatic Birds, Russia, 2016. Emerg. Infect. Dis. 2017, 23, 359–360. [Google Scholar] [CrossRef]
- Shin, J.; Kang, S.; Byeon, H.; Cho, S.M.; Kim, S.Y.; Chung, Y.J.; Jung, S.H. Highly pathogenic H5N6 avian influenza virus subtype clade 2.3.4.4 indigenous in South Korea. Sci. Rep. 2020, 10, 7241. [Google Scholar] [CrossRef]
- Yamaji, R.; Saad, M.D.; Davis, C.T.; Swayne, D.E.; Wang, D.; Wong, F.Y.; McCauley, J.W.; Peiris, J.M.; Webby, R.J.; Fouchier, R.A.; et al. Pandemic potential of highly pathogenic avian influenza clade 2.3.4.4 A(H5) viruses. Rev. Med. Virol. 2020, 30, e2099. [Google Scholar] [CrossRef]
- Stallknecht, D.E.; Kienzle-Dean, C.; Davis-Fields, N.; Jennelle, C.S.; Bowman, A.S.; Nolting, J.M.; Boyce, W.M.; Crum, J.M.; Santos, J.J.; Brown, J.D.; et al. Limited detection of antibodies to clade 2.3.4.4 a/goose/guangdong/1/1996 lineage Highly Pathogenic H5 Avian Influenza virus in North American waterfowl. J. Wildl. Dis. 2020, 56, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Pavon-Jordan, D. Waterbirds in a Changing World: Effects of Climate, Habitat, and Conservation Policy on European Wa-terbirds. Ph.D. Thesis, Norwegian Institute for Nature Research, Trondheim, Norway, 2017. [Google Scholar]
- Verhagen, J.H.; Fouchier, R.A.M.; Lewis, N. Highly Pathogenic Avian Influenza Viruses at the Wild–Domestic Bird Interface in Europe: Future Directions for Research and Surveillance. Viruses 2021, 13, 212. [Google Scholar] [CrossRef]
- Caliendo, V.; Lewis, N.S.; Pohlmann, A.; Baillie, S.R.; Banyard, A.C.; Beer, M.; Brown, I.H.; Fouchier, R.A.M.; Hansen, R.D.E.; Lameris, T.K.; et al. Transatlantic spread of highly pathogenic avian influenza H5N1 by wild birds from Europe to North America in 2021. Sci. Rep. 2022, 12, 11729. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Müller, N.F.; Bouckaert, R.; Xu, B.; Drummond, A.J. Bayesian phylodynamics of avian influenza A virus H9N2 in Asia with time-dependent predictors of migration. PLOS Comput. Biol. 2019, 15, e1007189. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xie, D.; Nie, Z.; Xu, B.; Drummond, A.J. Inferring host roles in bayesian phylodynamics of global avian influenza A virus H9N2. Virology 2019, 538, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.T.; Edwards, K.; Qiu, X.; Kim, D.-K.; Hixson, J.E.; Krauss, S.; Webby, R.J.; Webster, R.G.; Bahl, J. Host diversity and behavior determine patterns of interspecies transmission and geographic diffusion of avian influenza A subtypes among North American wild reservoir species. PLoS Pathog. 2022, 18, e1009973. [Google Scholar] [CrossRef]
- Magee, D.; Beard, R.; Suchard, M.A.; Lemey, P.; Scotch, M. Combining phylogeography and spatial epidemiology to uncover predictors of H5N1 influenza A virus diffusion. Arch. Virol. 2014, 160, 215–224. [Google Scholar] [CrossRef]
- Hicks, J.T.; Lee, D.-H.; Duvvuri, V.R.; Torchetti, M.K.; Swayne, D.E.; Bahl, J. Agricultural and geographic factors shaped the North American 2015 highly pathogenic avian influenza H5N2 outbreak. PLoS Pathog. 2020, 16, e1007857. [Google Scholar] [CrossRef]
- Dudas, G.; Carvalho, L.M.; Bedford, T.; Tatem, A.J.; Baele, G.; Faria, N.R.; Park, D.J.; Ladner, J.T.; Arias, A.; Asogun, D.; et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 2017, 544, 309–315. [Google Scholar] [CrossRef]
- Morin, C.W.; Stoner-Duncan, B.; Winker, K.; Scotch, M.; Hess, J.J.; Meschke, J.S.; Ebi, K.L.; Rabinowitz, P.M. Avian influenza virus ecology and evolution through a climatic lens. Environ. Int. 2018, 119, 241–249. [Google Scholar] [CrossRef]
- Gilbert, M.; Slingenbergh, J.; Xiao, X. Climate change and avian influenza. Rev. Sci. Tech. l’OIE 2008, 27, 459–466. [Google Scholar] [CrossRef]
- Vandegrift, K.J.; Sokolow, S.; Daszak, P.; Kilpatrick, A.M. Ecology of avian influenza viruses in a changing world. Ann. N. Y. Acad. Sci. 2010, 1195, 113–128. [Google Scholar] [CrossRef]
- Hinzman, L.D.; Bettez, N.D.; Bolton, W.R.; Chapin, F.S.; Dyurgerov, M.B.; Fastie, C.L.; Griffith, B.; Hollister, R.D.; Hope, A.; Huntington, H.P.; et al. Evidence and Implications of Recent Climate Change in Northern Alaska and Other Arctic Regions. Clim. Chang. 2005, 72, 251–298. [Google Scholar] [CrossRef]
- Arctic Monitoring and Assessment Programme (AMAP). AMAP Climate Change Update 2019: An Update to Key Findings of Snow, Water, Ice and Permafrost in the Arctic; Arctic Monitoring and Assessment Programme (AMAP): Oslo, Norway, 2019. [Google Scholar]
- Gass, J.D.; Dusek, R.J.; Hall, J.S.; Hallgrimsson, G.T.; Halldórsson, H.P.; Vignisson, S.R.; Ragnarsdottir, S.B.; Jónsson, J.E.; Krauss, S.; Wong, S.S.; et al. Global dissemination of Influenza A virus is driven by wild bird migration through arctic and subarctic zones Authorea. Mol. Ecol. 2022, 32, 198–213. [Google Scholar] [CrossRef] [PubMed]
- A Herrick, K.; Huettmann, F.; A Lindgren, M. A global model of avian influenza prediction in wild birds: The importance of northern regions. Vet. Res. 2013, 44, 42. [Google Scholar] [CrossRef]
- Hall, J.S.; Dusek, R.J.; Nashold, S.W.; TeSlaa, J.L.; Allen, R.B.; Grear, D.A. Avian influenza virus prevalence in marine birds is dependent on ocean temperatures. Ecol. Appl. 2019, 30, e02040. [Google Scholar] [CrossRef] [PubMed]
- Ferenczi, M.; Beckmann, C.; Warner, S.; Loyn, R.; O’riley, K.; Wang, X.; Klaassen, M. Avian influenza infection dynamics under variable climatic conditions, viral prevalence is rainfall driven in waterfowl from temperate, south-east Australia. Vet. Res. 2016, 47, 23. [Google Scholar] [CrossRef] [PubMed]
- Bahl, J.; Pham, T.T.; Hill, N.J.; Hussein, I.T.M.; Ma, E.J.; Easterday, B.C.; Halpin, R.A.; Stockwell, T.B.; Wentworth, D.E.; Kayali, G.; et al. Ecosystem Interactions Underlie the Spread of Avian Influenza A Viruses with Pandemic Potential. PLoS Pathog. 2016, 12, e1005620. [Google Scholar] [CrossRef]
- Rogers, S.O.; Starmer, W.T.; Castello, J.D. Recycling of pathogenic microbes through survival in ice. Med. Hypotheses 2004, 63, 773–777. [Google Scholar] [CrossRef]
- Kirby, J.S.; Stattersfield, A.J.; Butchart, S.H.M.; Evans, M.I.; Grimmett, R.F.A.; Jones, V.R.; O’Sullivan, J.; Tucker, G.M.; Newton, I. Key conservation issues for migratory land- and waterbird species on the world’s major flyways. Bird Conserv. Int. 2008, 18, S49–S73. [Google Scholar] [CrossRef]
- Bao, Y.; Bolotov, P.; Dernovoy, D.; Kiryutin, B.; Zaslavsky, L.; Tatusova, T.; Ostell, J.; Lipman, D. The Influenza Virus Resource at the National Center for Biotechnology Information. J. Virol. 2008, 82, 596–601. [Google Scholar] [CrossRef]
- Puryear, W.; Sawatzki, K.; Hill, N.; Foss, A.; Stone, J.J.; Doughty, L.; Walk, D.; Gilbert, K.; Murray, M.; Cox, E.; et al. Outbreak of Highly Pathogenic Avian Influenza H5N1 in New England Seals. bioRxiv 2022, 30, 2022-07. [Google Scholar]
- Hicks, J.T.; Dimitrov, K.M.; Afonso, C.L.; Ramey, A.M.; Bahl, J. Global phylodynamic analysis of avian paramyxovirus-1 provides evidence of inter-host transmission and intercontinental spatial diffusion. BMC Evol. Biol. 2019, 19, 108. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Tavaré, S.; Miura, R.M. Lectures on mathematics in the life sciences. Am. Math. Soc. 1986, 17, 57–86. [Google Scholar]
- Yang, Z. Maximum-likelihood models for combined analyses of multiple sequence data. J. Mol. Evol. 1996, 42, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Drummond, A.J.; Nicholls, G.K.; Rodrigo, A.; Solomon, W. Estimating Mutation Parameters, Population History and Genealogy Simultaneously From Temporally Spaced Sequence Data. Genetics 2002, 161, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Ayres, D.L.; Rambaut, A.; Suchard, M.A.; Lemey, P. High-Performance Computing in Bayesian Phylogenetics and Phylodynamics Using BEAGLE. In Evolutionary Genomics: Statistical and Computational Methods; Anisimova, M., Ed.; Springer: New York, NY, USA, 2019; pp. 691–722. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Pagel, M.; Meade, A.; Barker, D. Bayesian Estimation of Ancestral Character States on Phylogenies. Syst. Biol. 2004, 53, 673–684. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree; v 1.4. 4. 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 3 February 2022).
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Hong, S.L.; Lemey, P.; Suchard, M.A.; Baele, G. Bayesian Phylogeographic Analysis Incorporating Predictors and Individual Travel Histories in BEAST. Curr. Protoc. 2021, 1, e98. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D.; et al. Unifying Viral Genetics and Human Transportation Data to Predict the Global Transmission Dynamics of Human Influenza H3N2. PLoS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.; Magee, D.; Suchard, M.A.; Lemey, P.; Scotch, M. Generalized Linear Models for Identifying Predictors of the Evolutionary Diffusion of Viruses. AMIA Jt. Summits Transl. Sci. proceedings. AMIA Jt. Summits Transl. Sci. 2014, 2014, 23–28. [Google Scholar]
- Kuo, L.; Mallick, B. Variable Selection for Regression Models. Sankhyā Indian J. Stat. Ser. B 1998, 60, 65–81. [Google Scholar]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Duchatel, F.; Bronsvoort, B.M.D.C.; Lycett, S. Phylogeographic Analysis and Identification of Factors Impacting the Diffusion of Foot-and-Mouth Disease Virus in Africa. Front. Ecol. Evol. 2019, 7, 371. [Google Scholar] [CrossRef]
- Müller, N.F.; Dudas, G.; Stadler, T. Inferring time-dependent migration and coalescence patterns from genetic sequence and predictor data in structured populations. Virus Evol. 2019, 5, vez030. [Google Scholar] [CrossRef]
- Brown, V.L.; Rohani, P. The consequences of climate change at an avian influenza ‘hotspot’. Biol. Lett. 2012, 8, 1036–1039. [Google Scholar] [CrossRef]
- Harvell, D.; Altizer, S.; Cattadori, I.M.; Harrington, L.; Weil, E. Climate change and wildlife diseases: When does the host matter the most? Ecology 2009, 90, 912–920. [Google Scholar] [CrossRef]
- Banyard, A.C.; Lean, F.Z.X.; Robinson, C.; Howie, F.; Tyler, G.; Nisbet, C.; Seekings, J.; Meyer, S.; Whittard, E.; Ashpitel, H.F.; et al. Detection of Highly Pathogenic Avian Influenza Virus H5N1 Clade 2.3.4.4b in Great Skuas: A Species of Conservation Concern in Great Britain. Viruses 2022, 14, 212. [Google Scholar] [CrossRef]
- Shin, D.-L.; Siebert, U.; Lakemeyer, J.; Grilo, M.; Pawliczka, I.; Wu, N.-H.; Valentin-Weigand, P.; Haas, L.; Herrler, G. Highly Pathogenic Avian Influenza A(H5N8) Virus in Gray Seals, Baltic Sea. Emerg. Infect. Dis. 2019, 25, 2295–2298. [Google Scholar] [CrossRef]
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; Meruyert, S.; King, J.; Harder, T.; et al. Emergence and spread of novel H5N8, H5N5 and H5N1 clade 2.3.4.4 highly pathogenic avian influenza in 2020. Emerg. Microbes Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef]
- Wille, M.; Robertson, G.; Whitney, H.; Ojkic, D.; Lang, A.S. Reassortment of American and Eurasian genes in an influenza A virus isolated from a great black-backed gull (Larus marinus), a species demonstrated to move between these regions. Arch. Virol. 2010, 156, 107–115. [Google Scholar] [CrossRef]
- Dusek, R.J.; Hallgrimsson, G.T.; Ip, H.; Jónsson, J.E.; Sreevatsan, S.; Nashold, S.W.; TeSlaa, J.; Enomoto, S.; Halpin, R.A.; Lin, X.; et al. North Atlantic Migratory Bird Flyways Provide Routes for Intercontinental Movement of Avian Influenza Viruses. PLoS ONE 2014, 9, e92075. [Google Scholar] [CrossRef] [PubMed]
- Bevins, S.N.; Shriner, S.A.; Cumbee, J.C.; Dilione, K.E.; Douglass, K.E.; Ellis, J.W.; Killian, M.L.; Torchetti, M.K.; Lenoch, J.B. Intercontinental Movement of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4 Virus to the United States, 2021. Emerg. Infect. Dis. 2022, 28, 1006–1011. [Google Scholar] [CrossRef]
- Viboud, C.; Bjørnstad, O.N.; Smith, D.L.; Simonsen, L.; Miller, M.A.; Grenfell, B.T. Synchrony, Waves, and Spatial Hierarchies in the Spread of Influenza. Science 2006, 312, 447–451. [Google Scholar] [CrossRef]
- Wenger, J.B.; Naumova, E.N. Seasonal Synchronization of Influenza in the United States Older Adult Population. PLoS ONE 2010, 5, e10187. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, E.; Wenger, J.B.; Fefferman, N.H.; Bina, D.; Gradus, S.; Bhattacharyya, S.; Naumov, Y.N.; Gorski, J.; Naumova, E. Disproportional effects in populations of concern for pandemic influenza: Insights from seasonal epidemics in Wisconsin, 1967-2004. Influ. Other Respir. Viruses 2010, 4, 205–212. [Google Scholar] [CrossRef]
- Chui, K.K.H.; Cohen, S.A.; Naumova, E.N. Snowbirds and infection--new phenomena in pneumonia and influenza hospi-talizations from winter migration of older adults: A spatiotemporal analysis. BMC Public Health 2011, 11, 444. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.J.; Bishop, M.A.; Trovão, N.S.; Ineson, K.M.; Schaefer, A.L.; Puryear, W.B.; Zhou, K.; Foss, A.D.; Clark, D.E.; MacKenzie, K.G.; et al. Ecological divergence of wild birds drives avian influenza spillover and global spread. PLoS Pathog. 2022, 18, e1010062. [Google Scholar] [CrossRef]
- Cornell Lab of Ornithology. Species of Birds in North America. 2021. Available online: https://www.birds.cornell.edu/home/ (accessed on 3 February 2022).
- BirdLife International, European Red List of Birds 2021. 2021. Available online: https://www.iucnredlist.org/resources/birdlife2021 (accessed on 3 February 2022).
- Stallknecht, D.E.; Brown, J.D. Ecology of Avian Influenza in Wild Birds. Avian Influenza 2008, 1, 43–58. [Google Scholar] [CrossRef]
- Fourment, M.; Darling, A.E.; Holmes, E.C. The impact of migratory flyways on the spread of avian influenza virus in North America. BMC Evol. Biol. 2017, 17, 118. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, J.G.; Verhagen, J.H.; Wille, M.; Waldenström, J. Host and virus ecology as determinants of influenza A virus transmission in wild birds. Curr. Opin. Virol. 2018, 28, 26–36. [Google Scholar] [CrossRef]
- Gallana, M.; Ryser-Degiorgis, M.-P.; Wahli, T.; Segner, H. Climate change and infectious diseases of wildlife: Altered interactions between pathogens, vectors and hosts. Curr. Zool. 2013, 59, 427–437. [Google Scholar] [CrossRef]
- Brown, J.D.; Goekjian, G.; Poulson, R.; Valeika, S.; Stallknecht, D.E. Avian influenza virus in water: Infectivity is dependent on pH, salinity and temperature. Vet. Microbiol. 2009, 136, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Stallknecht, D.E.; Shane, S.M. Host range of avian influenza virus in free-living birds. Vet. Res. Commun. 1988, 12, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Paek, M.; Lee, Y.; Yoon, H.; Kang, H.; Kim, M.; Choi, J.; Jeong, O.; Kwon, J.; Moon, O.; Lee, S. Survival rate of H5N1 highly pathogenic avian influenza viruses at different temperatures. Poult. Sci. 2010, 89, 1647–1650. [Google Scholar] [CrossRef]
- Kurmi, B.; Murugkar, H.V.; Nagarajan, S.; Tosh, C.; Dubey, S.C.; Kumar, M. Survivability of Highly Pathogenic Avian Influenza H5N1 Virus in Poultry Faeces at Different Temperatures. Indian J. Virol. 2013, 24, 272–277. [Google Scholar] [CrossRef]
- Coombe, M.; Iwasawa, S.; Byers, K.A.; Prystajecky, N.; Hsiao, W.; Patrick, D.M.; Himsworth, C.G. A Systematic Review and Narrative Synthesis of the Use of Environmental Samples for the Surveillance of Avian Influenza Viruses in Wild Waterbirds. J. Wildl. Dis. 2021, 57, 1–18. [Google Scholar] [CrossRef]
- Tingley, M.W.; Koo, M.; Moritz, C.; Rush, A.C.; Beissinger, S.R. The push and pull of climate change causes heterogeneous shifts in avian elevational ranges. Glob. Chang. Biol. 2012, 18, 3279–3290. [Google Scholar] [CrossRef]
- Magge, A.; Weissenbacher, D.; O’Connor, K.; Tahsin, T.; Gonzalez-Hernandez, G.; Scotch, M. GeoBoost2: A natural language processing pipeline for GenBank metadata enrichment for virus phyloge-ography. Bioinformatics 2020, 36, 5120–5121. [Google Scholar] [CrossRef] [PubMed]
- Gaskell, D.E.; Huber, M.; O’brien, C.L.; Inglis, G.N.; Acosta, R.P.; Poulsen, C.J.; Hull, P.M. The latitudinal temperature gradient and its climate dependence as inferred from foraminiferal δ 18 O over the past 95 million years. Proc. Natl. Acad. Sci. USA 2022, 119, e2111332119. [Google Scholar] [CrossRef] [PubMed]
- Alkie, T.N.; Lopes, S.; Hisanaga, T.; Xu, W.; Suderman, M.; Koziuk, J.; Fisher, M.; Redford, T.; Lung, O.; Joseph, T.; et al. A threat from both sides: Multiple introductions of genetically distinct H5 HPAI viruses into Canada via both East Asia-Australasia/Pacific and Atlantic flyways. Virus Evol. 2022, 8, veac077. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Predictor | Justification | Value | Data Source |
|---|---|---|---|
| Distance between centroids | Decreased distance between states has been shown to relate to viral spread between geographic states in several phylogeographic-GLM models [15,16,17,45]. | Great circle distance in kilometers (km) between geographic state centroids | Google Earth |
| Latitude | Given that many migratory avian species breed in northern latitudes, higher latitudes may increase global dispersal of IAVs due to transmission dynamics between adults and juveniles at breeding ranges, spreading viruses globally [13,15,16,17,24]. | Decimal latitude at exact centroid of geographic region | Google Earth |
| Shared borders | Shared borders have been implicated in the spread of viruses between both humans and animals due to geographic proximity and movement patterns [13,50]. | Binary 0/1 (no/yes) | Google Earth |
| Northward movement | Northward movement has been associated with global spread of IAVs due to the condensed land masses around the circumpolar perimeter of the Arctic that connect hemispheres, particularly following breeding season [10,11]. Northward movement has not been previously used as a predictor in standard GLMs. | Binary 0/1 (no/yes) whether geographic state of origin is north by latitude of state of destination | Google Earth |
| Precipitation | Precipitation has been modeled in GLMs previously, with varying significance [13,15,16,17,49]. | Mean yearly precipitation in mm, 2020 | US: NOAA National Centers for Environmental Information; Europe: The World Bank Open Data |
| Change in precipitation | Change in precipitation has not been previously used as a predictor in standard GLMs. | Difference between mean precipitation of 1901–2000 and that of 2020 | US: NOAA National Centers for Environmental Information; Europe: The World Bank Open Data, The World Bank Group Climate Change Knowledge Portal |
| Air temperature | Air temperature has been modeled in GLMs previously, with varying significance [13,15,16,18,49]. | Mean yearly air temperature in Celsius, 2020 | US: NOAA National Centers for Environmental Information; Europe: The World Bank Open Data |
| Change in air temperature | Change in air temperature has not been previously used as a predictor in standard GLMs. | Difference between mean temperature of 1901–2000 and that of 2020 | US: NOAA National Centers for Environmental Information; Europe: The World Bank Open Data, The World Bank Group Climate Change Knowledge Portal |
| Sample size | Commonly included in GLMs to account for different sample sizes by state, which can bias results. GLM assumes that the sample sizes across subpopulations are proportional to the subpopulation sizes [13]. | Count of virus sequences from each geographic state | Downsampled dataset |
| Continental location of sequence: Europe or North America | There may be geographic differences in virus diffusion due to continental land size, proximity to nearby regions, or composition of host diversity and abundance in given regions. This has not been modeled previously. | Binary (0/1) | Downsampled dataset metadata |
| Variable Name | Mean | Standard Deviation | Range |
|---|---|---|---|
| Distance between centroids (km) | 4587.3 | 3101.5 | 690–7564 |
| Precipitation (mm) | 838.2 | 262.8 | 473.8–1163.2 |
| Precipitation change (mm) | 34.8 | 38.2 | −2.35–108.0 |
| Temperature (°C) | 10.13 | 2.66 | 7.60–14.39 |
| Temperature change (°C) | 1.37 | 0.092 | 1.23–1.48 |
| Latitude | 43.7 | 5.9 | 34.9–52.9 |
| Sample size (count) | 50.3 | 15.78 | 33–80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gass, J.D., Jr.; Hill, N.J.; Damodaran, L.; Naumova, E.N.; Nutter, F.B.; Runstadler, J.A. Ecogeographic Drivers of the Spatial Spread of Highly Pathogenic Avian Influenza Outbreaks in Europe and the United States, 2016–Early 2022. Int. J. Environ. Res. Public Health 2023, 20, 6030. https://doi.org/10.3390/ijerph20116030
Gass JD Jr., Hill NJ, Damodaran L, Naumova EN, Nutter FB, Runstadler JA. Ecogeographic Drivers of the Spatial Spread of Highly Pathogenic Avian Influenza Outbreaks in Europe and the United States, 2016–Early 2022. International Journal of Environmental Research and Public Health. 2023; 20(11):6030. https://doi.org/10.3390/ijerph20116030
Chicago/Turabian StyleGass, Jonathon D., Jr., Nichola J. Hill, Lambodhar Damodaran, Elena N. Naumova, Felicia B. Nutter, and Jonathan A. Runstadler. 2023. "Ecogeographic Drivers of the Spatial Spread of Highly Pathogenic Avian Influenza Outbreaks in Europe and the United States, 2016–Early 2022" International Journal of Environmental Research and Public Health 20, no. 11: 6030. https://doi.org/10.3390/ijerph20116030