
Gulf War Illness Is Associated with Host Gut Microbiome Dysbiosis and Is Linked to Altered Species Abundance in Veterans from the BBRAIN Cohort

 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. IRB Approval and Informed Consent
2.1.1. Gut Microbiome Analysis
DNA Extraction and Quantification
Library Preparation and Sequencing Methods
Bioinformatics Analysis Methods
Figure Generation and Statistical Analysis
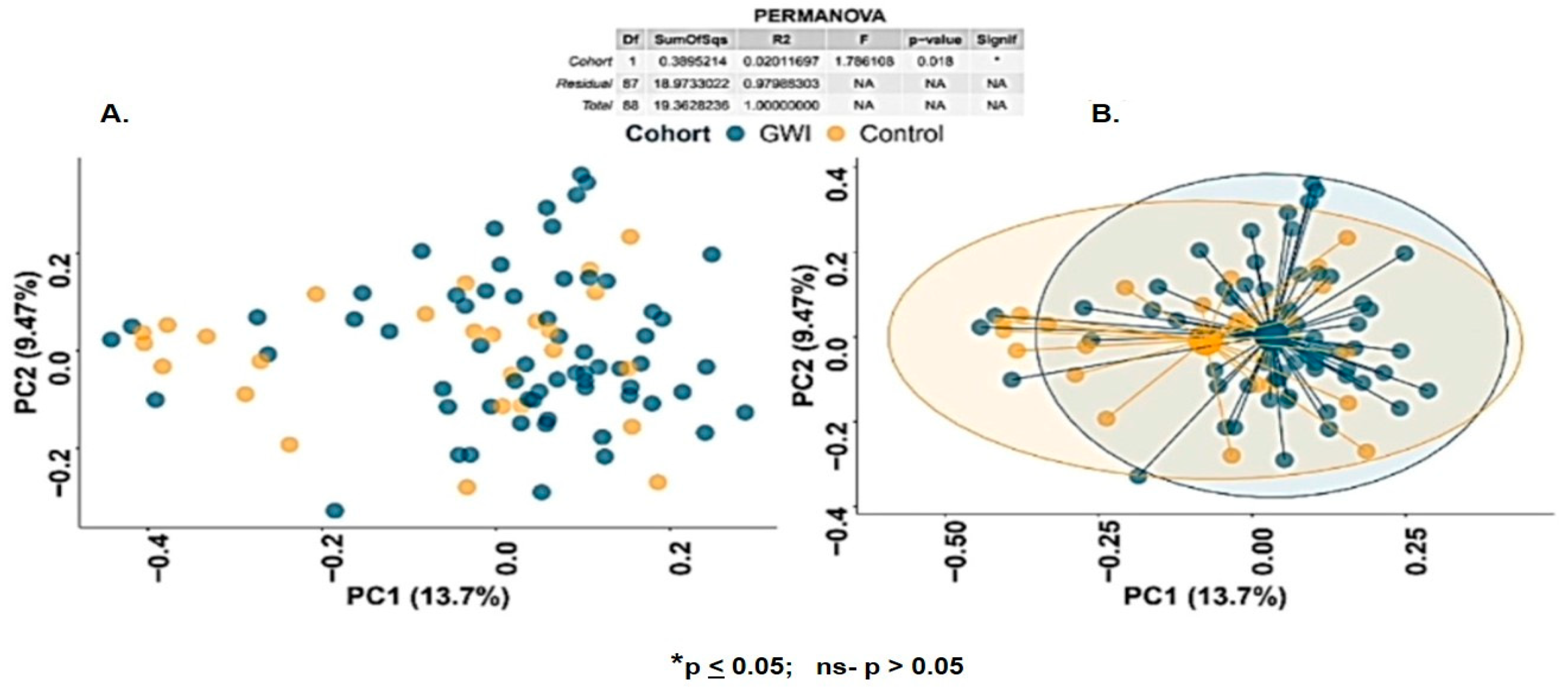
3. Results
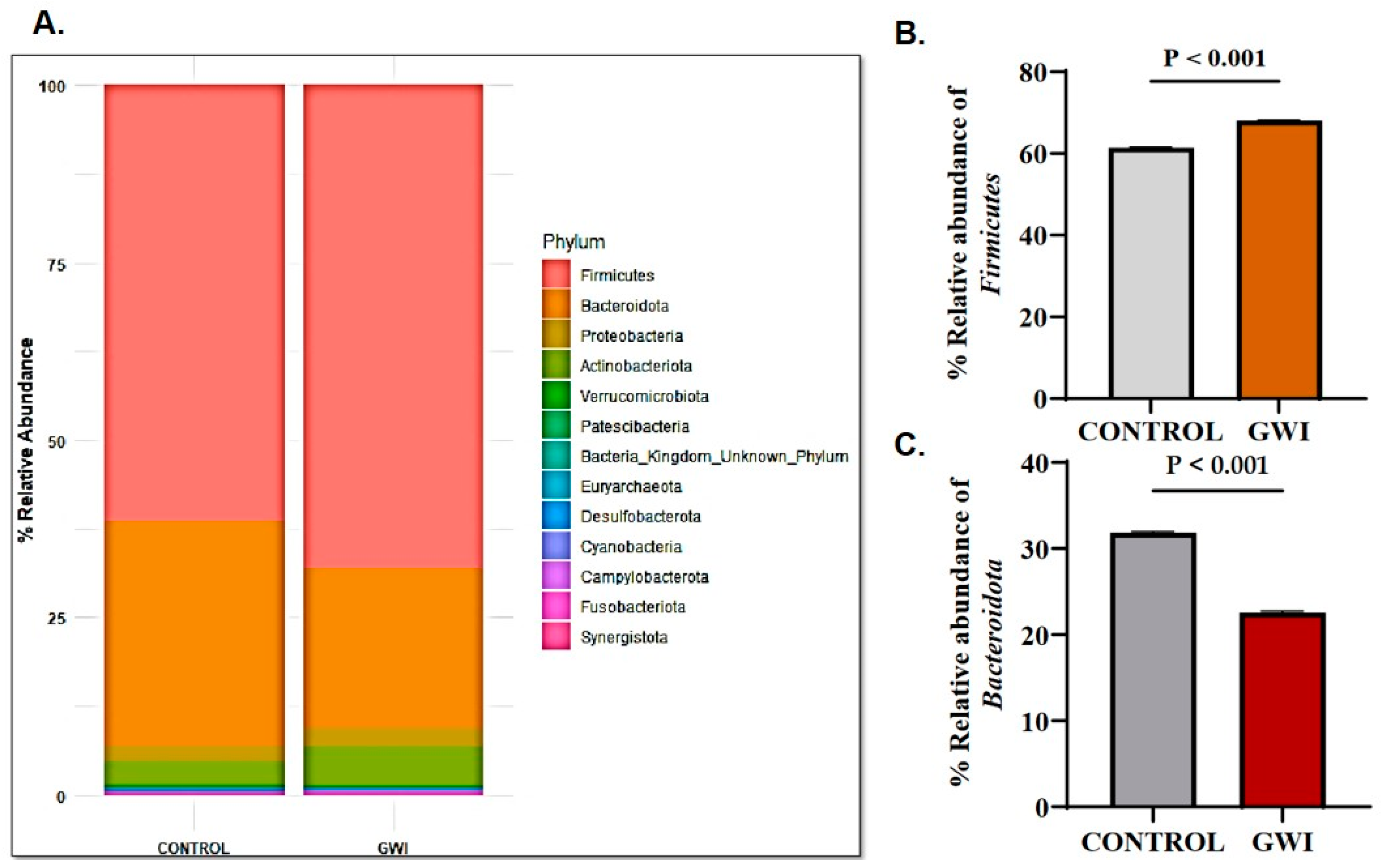
3.1. GWI Resulted in an Altered Firmicutes–Bacteroidetes Ratio and Expression of Unique Family and Genus Abundance
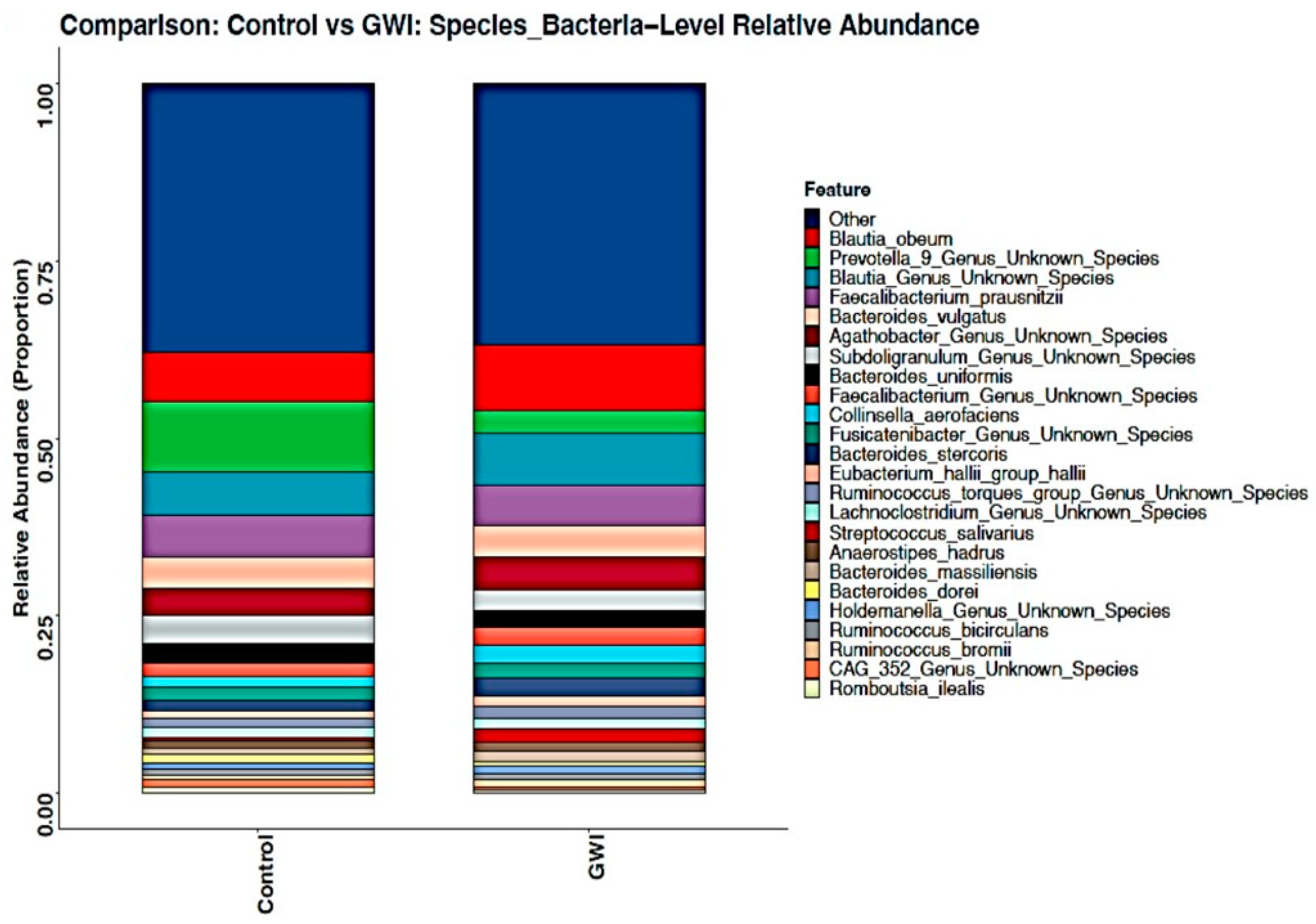
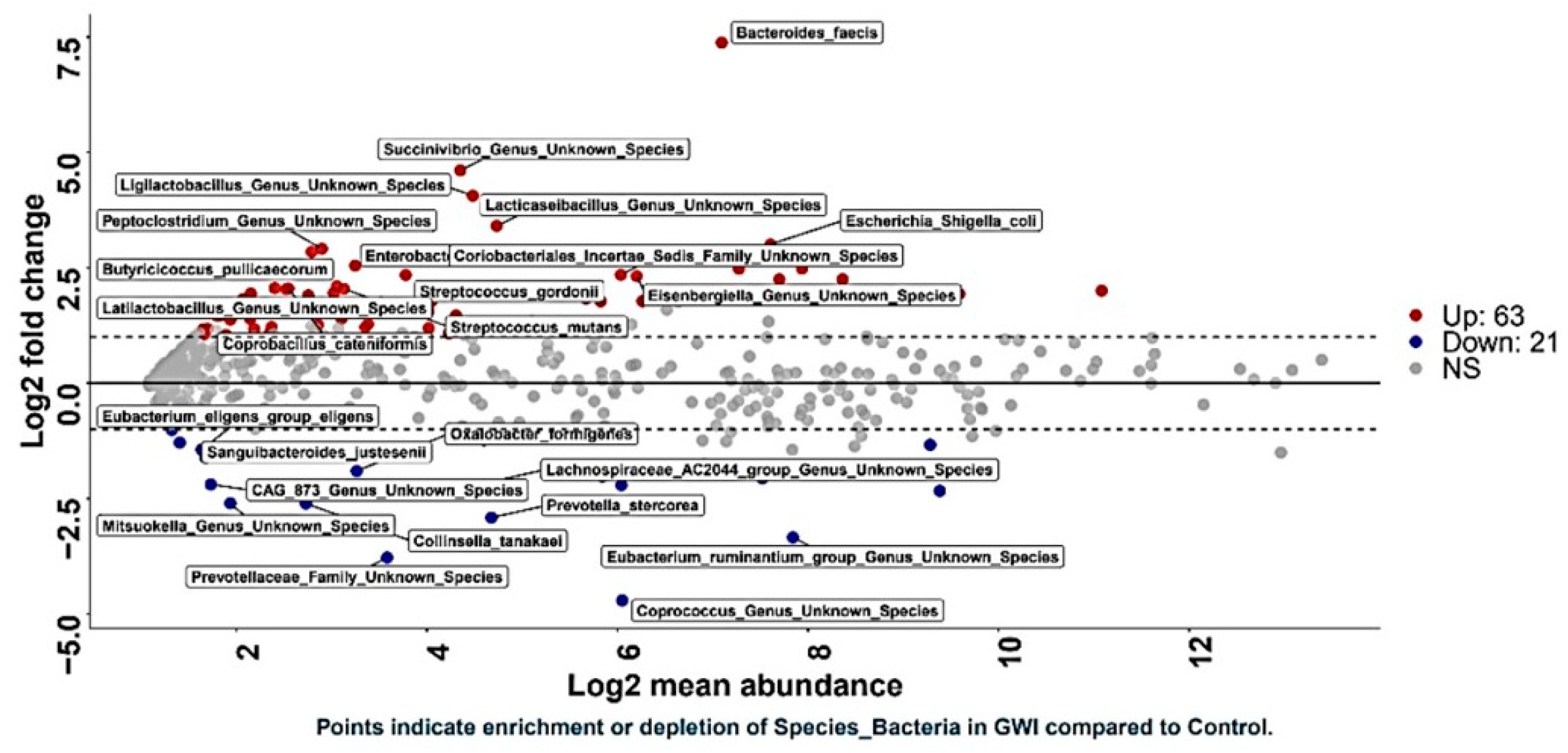
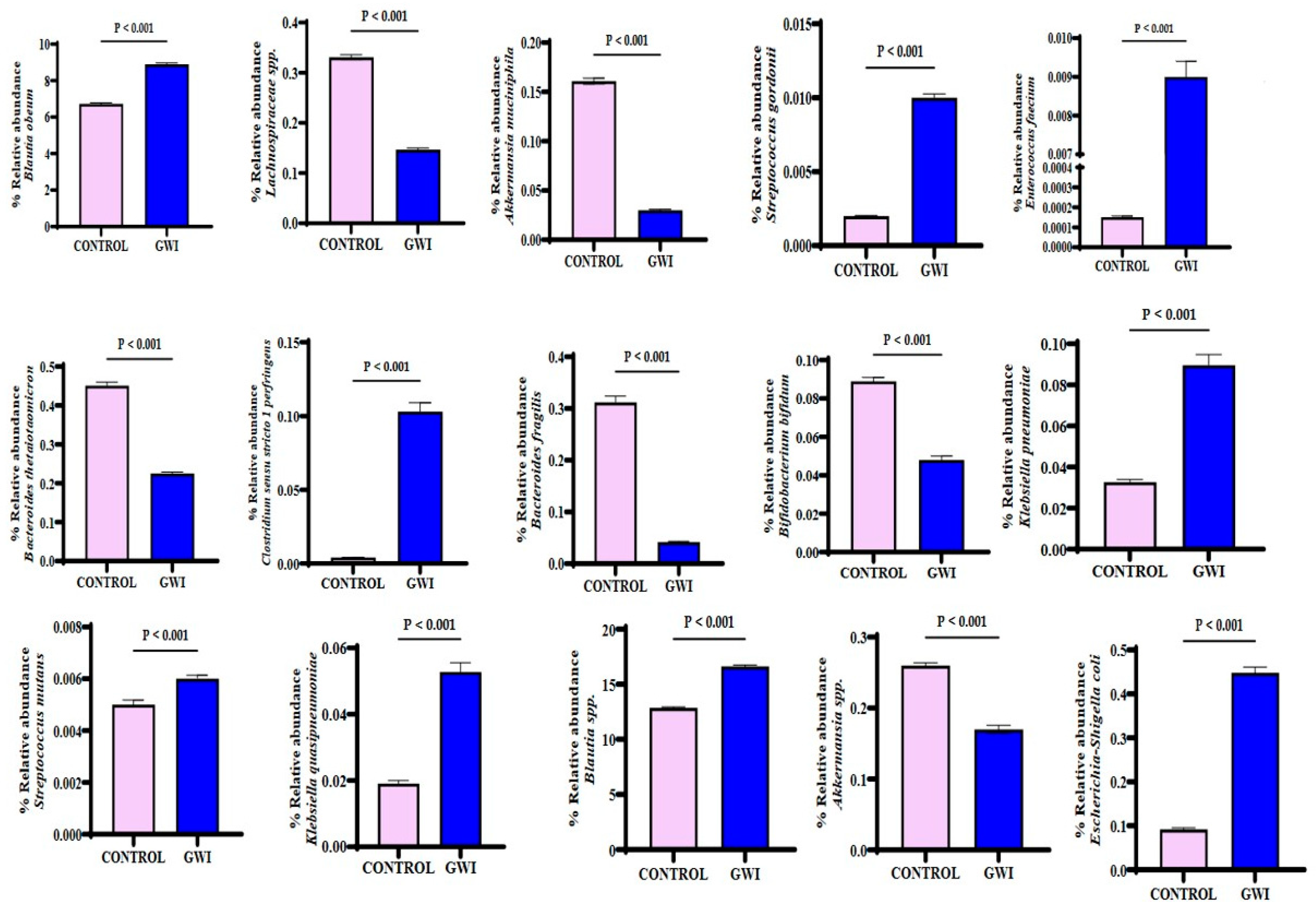
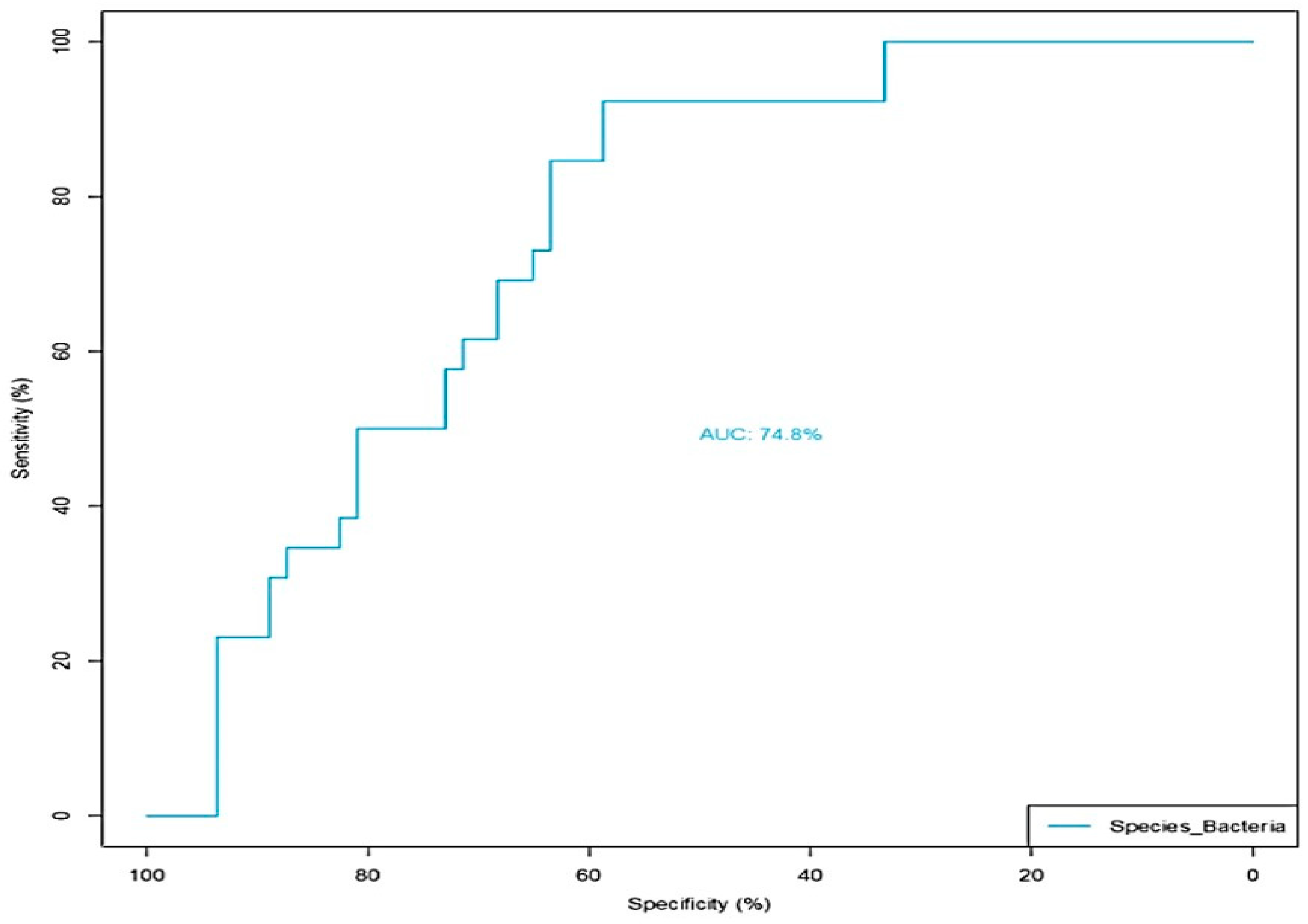
3.2. GWI Cases Expressed a Differential Abundance of Host Species While Presenting a Unique Species Signature Compared to Non-GWI Controls
3.3. GWI Cases Presented a Unique Bacterial Enzyme Expression Profile with a Differential Abundance of Peptidylprolyl Isomerase and NADH Ubiquinone Reductase, a Mitochondrial Respiration Enzyme
3.4. GWI Cases Presented a Unique Bacterial Biochemical Pathway That Was Influenced by Microbiome Profile
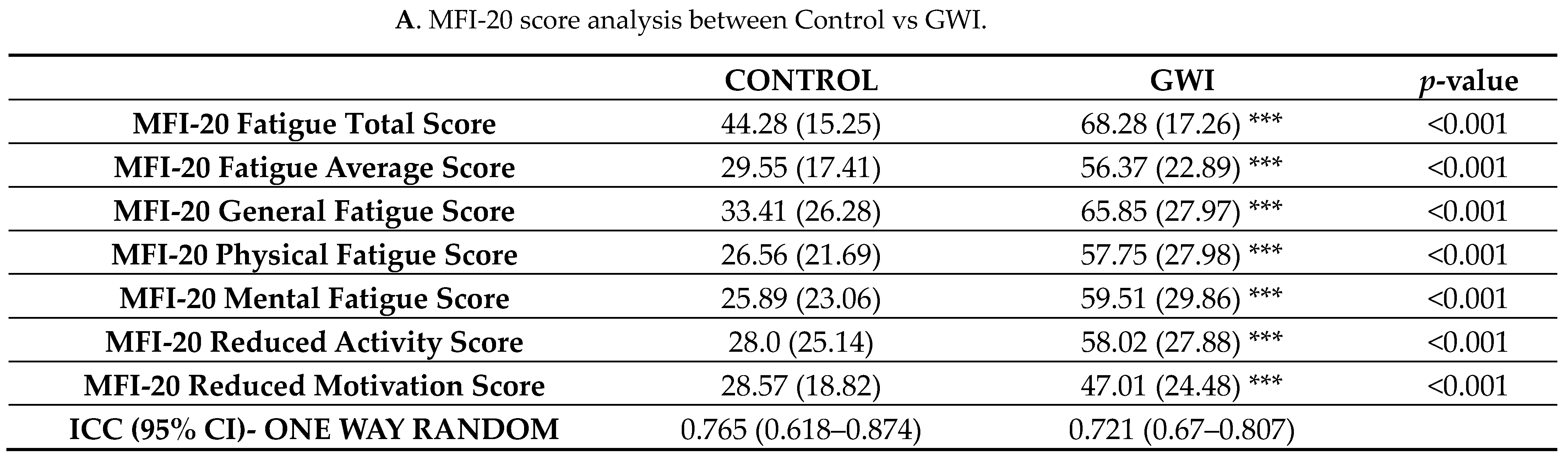
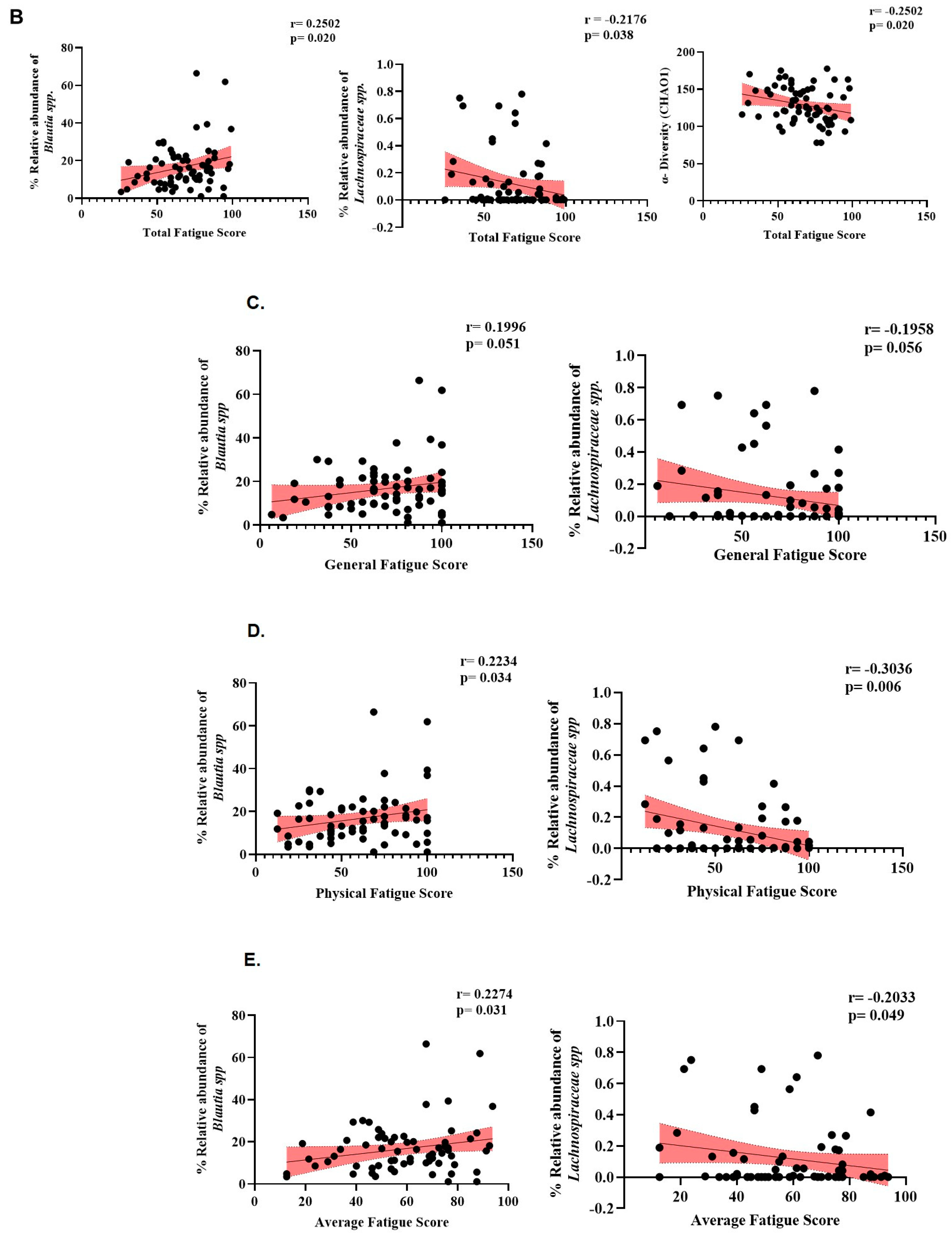
3.5. GWI Cases Altered Microbiome Diversity and Species Abundance Correlated with MFI Fatigue Scores
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Disclaimer
References
- Mawson, A.R.; Croft, A.M. Gulf War Illness: Unifying Hypothesis for a Continuing Health Problem. Int. J. Environ. Res. Public Health 2019, 16, 111. [Google Scholar] [CrossRef]
- White, R.F.; Steele, L.; O’Callaghan, J.P.; Sullivan, K.; Binns, J.H.; Golomb, B.A.; Bloom, F.E.; Bunker, J.A.; Crawford, F.; Graves, J.C.; et al. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: Effects of toxicant exposures during deployment. Cortex 2016, 74, 449–475. [Google Scholar] [CrossRef]
- Cohen, J.; Mathew, A.; Dourvetakis, K.D.; Sanchez-Guerrero, E.; Pangeni, R.P.; Gurusamy, N.; Aenlle, K.K.; Ravindran, G.; Twahir, A.; Isler, D.; et al. Recent Research Trends in Neuroinflammatory and Neurodegenerative Disorders. Cells 2024, 13, 511. [Google Scholar] [CrossRef]
- Golomb, B.A. Acetylcholinesterase inhibitors and Gulf War illnesses. Proc. Natl. Acad. Sci. USA 2008, 105, 4295–4300. [Google Scholar] [CrossRef] [PubMed]
- Alhasson, F.; Das, S.; Seth, R.; Dattaroy, D.; Chandrashekaran, V.; Ryan, C.N.; Chan, L.S.; Testerman, T.; Burch, J.; Hofseth, L.J.; et al. Altered gut microbiome in a mouse model of Gulf War Illness causes neuroinflammation and intestinal injury via leaky gut and TLR4 activation. PLoS ONE 2017, 12, e0172914. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bose, D.; Seth, R. Host gut microbiome and potential therapeutics in Gulf War Illness: A short review. Life Sci. 2021, 280, 119717. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.K.; Kimono, D.; Alhasson, F.; Sarkar, S.; Albadrani, M.; Lasley, S.K.; Horner, R.; Janulewicz, P.; Nagarkatti, M.; Nagarkatti, P.; et al. Increased butyrate priming in the gut stalls microbiome associated-gastrointestinal inflammation and hepatic metabolic reprogramming in a mouse model of Gulf War Illness. Toxicol. Appl. Pharmacol. 2018, 350, 64–77. [Google Scholar] [CrossRef]
- Kimono, D.; Sarkar, S.; Albadrani, M.; Seth, R.; Bose, D.; Mondal, A.; Li, Y.; Kar, A.N.; Nagarkatti, M.; Nagarkatti, P.; et al. Dysbiosis-Associated Enteric Glial Cell Immune-Activation and Redox Imbalance Modulate Tight Junction Protein Expression in Gulf War Illness Pathology. Front. Physiol. 2019, 10, 1229. [Google Scholar] [CrossRef]
- Seth, R.K.; Maqsood, R.; Mondal, A.; Bose, D.; Kimono, D.; Holland, L.A.; Lloyd, P.J.; Klimas, N.; Horner, R.D.; Sullivan, K.; et al. Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness. Viruses 2019, 11, 968. [Google Scholar] [CrossRef]
- Janulewicz, P.A.; Seth, R.K.; Carlson, J.M.; Ajama, J.; Quinn, E.; Heeren, T.; Klimas, N.; Lasley, S.M.; Horner, R.D.; Sullivan, K.; et al. The Gut-Microbiome in Gulf War Veterans: A Preliminary Report. Int. J. Environ. Res. Public Health 2019, 16, 3751. [Google Scholar] [CrossRef]
- Kimono, D.; Bose, D.; Seth, R.K.; Mondal, A.; Saha, P.; Janulewicz, P.; Sullivan, K.; Lasley, S.; Horner, R.; Klimas, N.; et al. Host Akkermansia muciniphila Abundance Correlates with Gulf War Illness Symptom Persistence via NLRP3-Mediated Neuroinflammation and Decreased Brain-Derived Neurotrophic Factor. Neurosci. Insights 2020, 15, 2633105520942480. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Mondal, A.; Saha, P.; Kimono, D.; Sarkar, S.; Seth, R.K.; Janulewicz, P.; Sullivan, K.; Horner, R.; Klimas, N.; et al. TLR Antagonism by Sparstolonin B Alters Microbial Signature and Modulates Gastrointestinal and Neuronal Inflammation in Gulf War Illness Preclinical Model. Brain Sci. 2020, 10, 532. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. N. Am. 2017, 46, 77–89. [Google Scholar] [CrossRef]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Boyle, S.H.; Gifford, E.J.; Sullivan, B.A.; Wenker, T.H.N.; Abs, N.; Ahmed, S.T.; Upchurch, J.; Vahey, J.; Stafford, C.; et al. Self-reported gastrointestinal disorders among veterans with gulf war illness with and without posttraumatic stress disorder. Neurogastroenterol. Motil. 2023, 35, e14548. [Google Scholar] [CrossRef]
- Collier, C.A.; Salikhova, A.; Sabir, S.; Foncerrada, S.; Raghavan, S.A. Crisis in the gut: Navigating gastrointestinal challenges in Gulf War Illness with bioengineering. Mil. Med. Res. 2024, 11, 45. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Segal, J.P.; Carding, S.R.; Hart, A.L.; Hold, G.L. The gut virome: The ‘missing link’ between gut bacteria and host immunity? Ther. Adv. Gastroenterol. 2019, 12, 1756284819836620. [Google Scholar] [CrossRef]
- Keating, D.; Zundel, C.; Abreu, M.; Krengel, M.; Aenlle, K.; Nichols, M.D.; Toomey, R.; Chao, L.; Golier, J.; Abdullah, L.; et al. Boston biorepository, recruitment and integrative network (BBRAIN): A resource for the Gulf War Illness scientific community. Life Sci. 2021, 284, 119903. [Google Scholar] [CrossRef] [PubMed]
- Steele, L. Prevalence and patterns of Gulf War illness in Kansas veterans: Association of symptoms with characteristics of person, place, and time of military service. Am. J. Epidemiol. 2000, 152, 992–1002. [Google Scholar] [CrossRef]
- Sultana, E.; Shastry, N.; Kasarla, R.; Hardy, J.; Collado, F.; Aenlle, K.; Abreu, M.; Sisson, E.; Sullivan, K.; Klimas, N.; et al. Disentangling the effects of PTSD from Gulf War Illness in male veterans via a systems-wide analysis of immune cell, cytokine, and symptom measures. Mil. Med. Res. 2024, 11, 2. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Straub, D.; Blackwell, N.; Langarica-Fuentes, A.; Peltzer, A.; Nahnsen, S.; Kleindienst, S. Interpretations of Environmental Microbial Community Studies Are Biased by the Selected 16S rRNA (Gene) Amplicon Sequencing Pipeline. Front. Microbiol. 2020, 11, 550420. [Google Scholar] [CrossRef]
- Li, M.; Kopylova, E.; Mao, J.; Namkoong, J.; Sanders, J.; Wu, J. Microbiome and lipidomic analysis reveal the interplay between skin bacteria and lipids in a cohort study. Front. Microbiol. 2024, 15, 1383656. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Karp, P.D.; Riley, M.; Paley, S.M.; Pellegrini-Toole, A. The MetaCyc Database. Nucleic Acids Res. 2002, 30, 59–61. [Google Scholar] [CrossRef]
- Zhao, H.; Sun, R.; Wu, L.; Huang, P.; Liu, W.; Ma, Q.; Liao, Q.; Du, J. Bioinformatics Identification and Experimental Validation of a Prognostic Model for the Survival of Lung Squamous Cell Carcinoma Patients. Biochem. Genet. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Willis, A.D. Rarefaction, Alpha Diversity, and Statistics. Front. Microbiol. 2019, 10, 2407. [Google Scholar] [CrossRef]
- Su, X. Elucidating the Beta-Diversity of the Microbiome: From Global Alignment to Local Alignment. mSystems 2021, 6, e0036321. [Google Scholar] [CrossRef] [PubMed]
- Santos-Marcos, J.A.; Perez-Jimenez, F.; Camargo, A. The role of diet and intestinal microbiota in the development of metabolic syndrome. J. Nutr. Biochem. 2019, 70, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Zhu, S.; Wang, B.; Duan, L. Alterations of Gut Microbiota in Patients with Irritable Bowel Syndrome Based on 16S rRNA-Targeted Sequencing: A Systematic Review. Clin. Transl. Gastroenterol. 2019, 10, e00012. [Google Scholar] [CrossRef]
- Hsu, Y.-J.; Huang, W.-C.; Lin, J.-S.; Chen, Y.-M.; Ho, S.-T.; Huang, C.-C.; Tung, Y.-T. Kefir Supplementation Modifies Gut Microbiota Composition, Reduces Physical Fatigue, and Improves Exercise Performance in Mice. Nutrients 2018, 10, 862. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, F.; Seifert, S.; Szymczak, S. Evaluation of variable selection methods for random forests and omics data sets. Brief Bioinform. 2019, 20, 492–503. [Google Scholar] [CrossRef]
- Guo, P.; Luo, Y.; Mai, G.; Zhang, M.; Wang, G.; Zhao, M.; Gao, L.; Li, F.; Zhou, F. Gene expression profile based classification models of psoriasis. Genomics 2014, 103, 48–55. [Google Scholar] [CrossRef]
- Saulnier, D.M.; Riehle, K.; Mistretta, T.A.; Diaz, M.A.; Mandal, D.; Raza, S.; Weidler, E.M.; Qin, X.; Coarfa, C.; Milosavljevic, A.; et al. Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology 2011, 141, 1782–1791. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Hoozemans, J.; de Brauw, M.; Nieuwdorp, M.; Gerdes, V. Gut Microbiome and Metabolites in Patients with NAFLD and after Bariatric Surgery: A Comprehensive Review. Metabolites 2021, 11, 353. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, T.; Seekatz, A.; Bassis, C.; Rhee, Y.; Yelin, R.D.; Fogg, L.; Dangana, T.; Cisneros, E.C.; A Weinstein, R.; Okamoto, K.; et al. Increased Relative Abundance of Klebsiella pneumoniae Carbapenemase-producing Klebsiella pneumoniae within the Gut Microbiota Is Associated with Risk of Bloodstream Infection in Long-term Acute Care Hospital Patients. Clin. Infect. Dis. 2019, 68, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.D.; Sun, N.; Canakis, A.; Park, W.Y.; Weber, H.C. Irritable Bowel Syndrome and the Gut Microbiome: A Comprehensive Review. J. Clin. Med. 2023, 12, 2558. [Google Scholar] [CrossRef] [PubMed]
- Piazzesi, A.; Putignani, L. Extremely small and incredibly close: Gut microbes as modulators of inflammation and targets for therapeutic intervention. Front. Microbiol. 2022, 13, 958346. [Google Scholar] [CrossRef]
- Wang, T.; Guo, X.K.; Xu, H. Disentangling the Progression of Non-alcoholic Fatty Liver Disease in the Human Gut Microbiota. Front. Microbiol. 2021, 12, 728823. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef]
- Pan, R.; Wang, L.; Xu, X.; Chen, Y.; Wang, H.; Wang, G.; Zhao, J.; Chen, W. Crosstalk between the Gut Microbiome and Colonic Motility in Chronic Constipation: Potential Mechanisms and Microbiota Modulation. Nutrients 2022, 14, 3704. [Google Scholar] [CrossRef]
- Szopinska-Tokov, J.; Dam, S.; Naaijen, J.; Konstanti, P.; Rommelse, N.; Belzer, C.; Buitelaar, J.; Franke, B.; Bloemendaal, M.; Aarts, E.; et al. Investigating the Gut Microbiota Composition of Individuals with Attention-Deficit/Hyperactivity Disorder and Association with Symptoms. Microorganisms 2020, 8, 406. [Google Scholar] [CrossRef]
- Togo, A.; Khelaifia, S.; Bittar, F.; Maraninchi, M.; Raoult, D.; Million, M. ‘Eisenbergiella massiliensis’, a new species isolated from human stool collected after bariatric surgery. New Microbes New Infect. 2016, 13, 15–16. [Google Scholar] [CrossRef]
- Liu, B.; Ye, D.; Yang, H.; Song, J.; Sun, X.; He, Z.; Mao, Y.; Hao, G. Assessing the relationship between gut microbiota and irritable bowel syndrome: A two-sample Mendelian randomization analysis. BMC Gastroenterol. 2023, 23, 150. [Google Scholar] [CrossRef]
- Sugimura, Y.; Kanda, A.; Sawada, K.; Wai, K.M.; Tanabu, A.; Ozato, N.; Midorikawa, T.; Hisada, T.; Nakaji, S.; Ihara, K. Association between Gut Microbiota and Body Composition in Japanese General Population: A Focus on Gut Microbiota and Skeletal Muscle. Int. J. Environ. Res. Public Health 2022, 19, 7464. [Google Scholar] [CrossRef] [PubMed]
- Goudman, L.; Demuyser, T.; Pilitsis, J.G.; Billot, M.; Roulaud, M.; Rigoard, P.; Moens, M. Gut dysbiosis in patients with chronic pain: A systematic review and meta-analysis. Front. Immunol. 2024, 15, 1342833. [Google Scholar] [CrossRef]
- Minerbi, A.; Fitzcharles, M.A. Gut microbiome: Pertinence in fibromyalgia. Clin. Exp. Rheumatol. 2020, 38, 99–104. [Google Scholar] [PubMed]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef]
- Tomasova, L.; Grman, M.; Ondrias, K.; Ufnal, M. The impact of gut microbiota metabolites on cellular bioenergetics and cardiometabolic health. Nutr. Metab. 2021, 18, 72. [Google Scholar] [CrossRef]
- Wolf, P.G.; Cowley, E.S.; Breister, A.; Matatov, S.; Lucio, L.; Polak, P.; Ridlon, J.M.; Gaskins, H.R.; Anantharaman, K. Diversity and distribution of sulfur metabolic genes in the human gut microbiome and their association with colorectal cancer. Microbiome 2022, 10, 64. [Google Scholar] [CrossRef]
- Buret, A.G.; Allain, T.; Motta, J.P.; Wallace, J.L. Effects of Hydrogen Sulfide on the Microbiome: From Toxicity to Therapy. Antioxid. Redox Signal. 2022, 36, 211–219. [Google Scholar] [CrossRef]
- Zhao, Z.; Xian, M.; Liu, M.; Zhao, G. Biochemical routes for uptake and conversion of xylose by microorganisms. Biotechnol. Biofuels 2020, 13, 21. [Google Scholar] [CrossRef]
- Hu, J.; Wu, Y.; Kang, L.; Liu, Y.; Ye, H.; Wang, R.; Zhao, J.; Zhang, G.; Li, X.; Wang, J.; et al. Dietary D-xylose promotes intestinal health by inducing phage production in Escherichia coli. NPJ Biofilms Microbiomes 2023, 9, 79. [Google Scholar] [CrossRef]
- Xiang, S.; Ye, K.; Li, M.; Ying, J.; Wang, H.; Han, J.; Shi, L.; Xiao, J.; Shen, Y.; Feng, X.; et al. Xylitol enhances synthesis of propionate in the colon via cross-feeding of gut microbiota. Microbiome 2021, 9, 62. [Google Scholar] [CrossRef]
- Golomb, B.A.; Han, J.H.; Fung, A.; Berg, B.K.; Miller, B.J.; Hamilton, G. Bioenergetic impairment in Gulf War illness assessed via (31)P-MRS. Sci. Rep. 2024, 14, 7418. [Google Scholar] [CrossRef]
- Sundberg, C.W.; Fitts, R.H. Bioenergetic basis of skeletal muscle fatigue. Curr. Opin. Physiol. 2019, 10, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Wylie, G.; Genova, H.; Dobryakova, E.; DeLuca, J.; Chiaravalloti, N.; Falvo, M.; Cook, D. Fatigue in Gulf War Illness is associated with tonically high activation in the executive control network. Neuroimage Clin. 2019, 21, 101641. [Google Scholar] [CrossRef] [PubMed]
- Boolani, A.; Gallivan, K.M.; Ondrak, K.S.; Christopher, C.J.; Castro, H.F.; Campagna, S.R.; Taylor, C.M.; Luo, M.; Dowd, S.E.; Smith, M.L.; et al. Trait Energy and Fatigue May Be Connected to Gut Bacteria among Young Physically Active Adults: An Exploratory Study. Nutrients 2022, 14, 466. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall | Case or Control Status | |||
|---|---|---|---|---|
| Control | GWI (Cases) | |||
| Total (N) | 89 | 26 | 63 | |
| 1. Age 2. Sex | Mean (Standard Deviation) 1: Male 2: Female | 57 (7) 71 (79.8%) 18 (20.2%) | 60 (10) 24 (92.3%) 2 (7.7%) | 56 (6) 47 (74.6%) 16 (25.4%) |
| 3. Hispanic or Latino descent | 1: Yes | 11 (12.8%) | 1 (4.0%) | 10 (16.4%) |
| 2: No | 75 (87.2%) | 24 (96.0%) | 51 (83.6%) | |
| 4. Race | 1: Black/African American | 9 (10.1%) | 2 (7.7%) | 7 (11.1%) |
| 2: White/Caucasian | 70 (78.7%) | 23 (88.5%) | 47 (74.6%) | |
| 3: Asian/Pacific Islander | 2 (2.2%) | 0 (0.0%) | 2 (3.2%) | |
| 4: Aleutian, Eskimo or American Indian | 2 (2.2%) | 1 (3.8%) | 1 (1.6%) | |
| 5: Other Multiracial | 6 (6.7%) | 0 (0.0%) | 6 (9.5%) | |
| 5. Total years of education | 15 (2) | 16 (2) | 15 (2) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trivedi, A.; Bose, D.; Moffat, K.; Pearson, E.; Walsh, D.; Cohen, D.; Skupsky, J.; Chao, L.; Golier, J.; Janulewicz, P.; et al. Gulf War Illness Is Associated with Host Gut Microbiome Dysbiosis and Is Linked to Altered Species Abundance in Veterans from the BBRAIN Cohort. Int. J. Environ. Res. Public Health 2024, 21, 1102. https://doi.org/10.3390/ijerph21081102
Trivedi A, Bose D, Moffat K, Pearson E, Walsh D, Cohen D, Skupsky J, Chao L, Golier J, Janulewicz P, et al. Gulf War Illness Is Associated with Host Gut Microbiome Dysbiosis and Is Linked to Altered Species Abundance in Veterans from the BBRAIN Cohort. International Journal of Environmental Research and Public Health. 2024; 21(8):1102. https://doi.org/10.3390/ijerph21081102
Chicago/Turabian StyleTrivedi, Ayushi, Dipro Bose, Kelly Moffat, Elisabeth Pearson, Dana Walsh, Devra Cohen, Jonathan Skupsky, Linda Chao, Julia Golier, Patricia Janulewicz, and et al. 2024. "Gulf War Illness Is Associated with Host Gut Microbiome Dysbiosis and Is Linked to Altered Species Abundance in Veterans from the BBRAIN Cohort" International Journal of Environmental Research and Public Health 21, no. 8: 1102. https://doi.org/10.3390/ijerph21081102
APA StyleTrivedi, A., Bose, D., Moffat, K., Pearson, E., Walsh, D., Cohen, D., Skupsky, J., Chao, L., Golier, J., Janulewicz, P., Sullivan, K., Krengel, M., Tuteja, A., Klimas, N., & Chatterjee, S. (2024). Gulf War Illness Is Associated with Host Gut Microbiome Dysbiosis and Is Linked to Altered Species Abundance in Veterans from the BBRAIN Cohort. International Journal of Environmental Research and Public Health, 21(8), 1102. https://doi.org/10.3390/ijerph21081102