Is Neurodegenerative Disease a Long-Latency Response to Early-Life Genotoxin Exposure?


Abstract
:1. Introduction
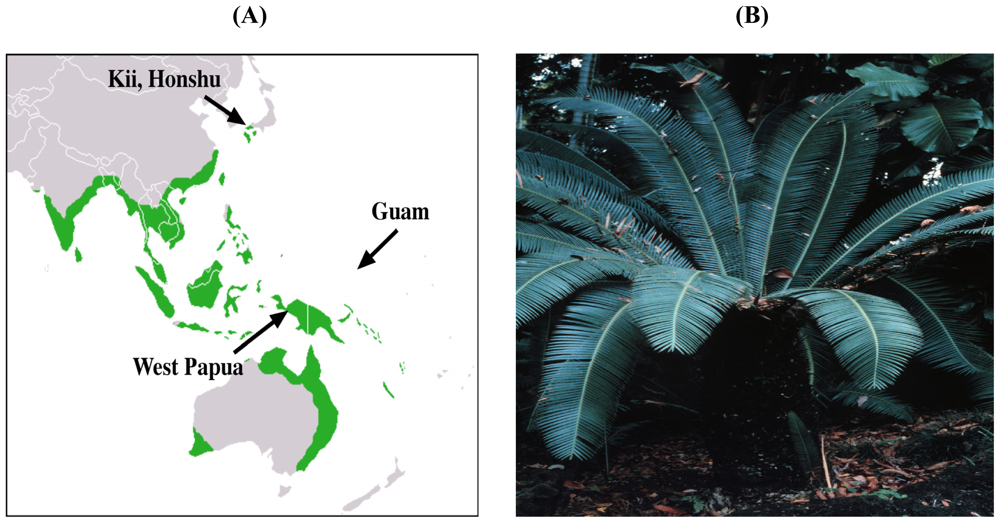
2. Evidence for the Role of Cycads in ALS-PDC
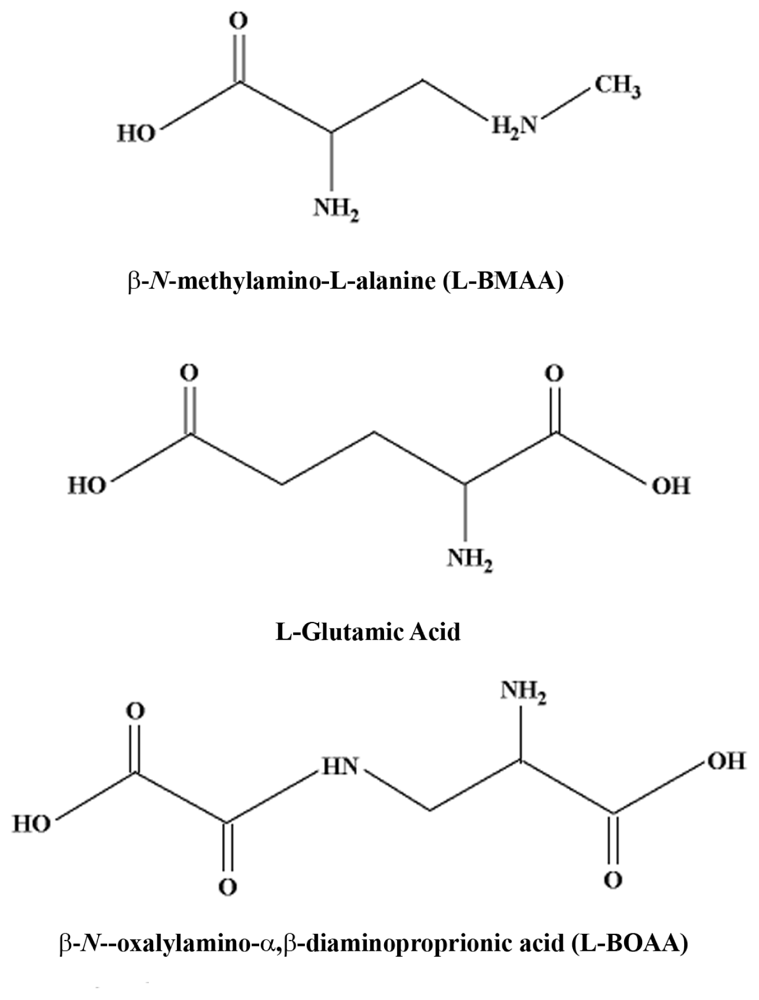
2.1. Cycad Toxins: β-N-Methylamino-L-Alanine (L-BMAA)
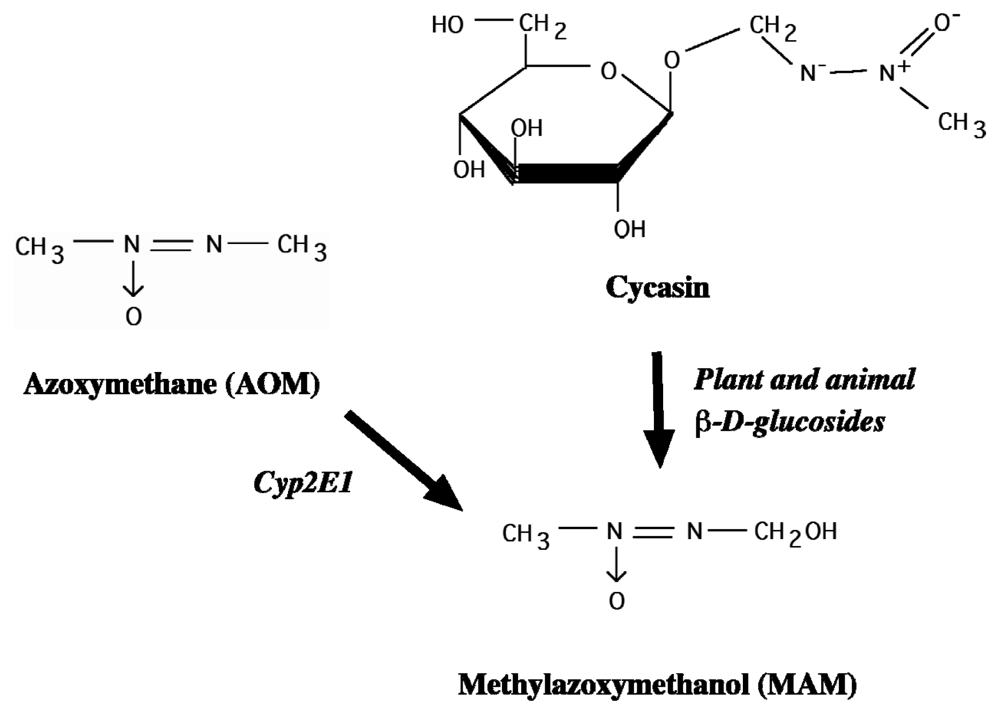
2.2. Cycad Toxins: Azoxyglycosides and Methylazoxymethanol (MAM)
3. Other Botanical Toxins, Toxicants and Neurodegenerative Disease
4. Cycasin and Brain Development
“Only small amounts of [cycad] are given to children because many become ill when they eat a dish made with cycad starch” [21].
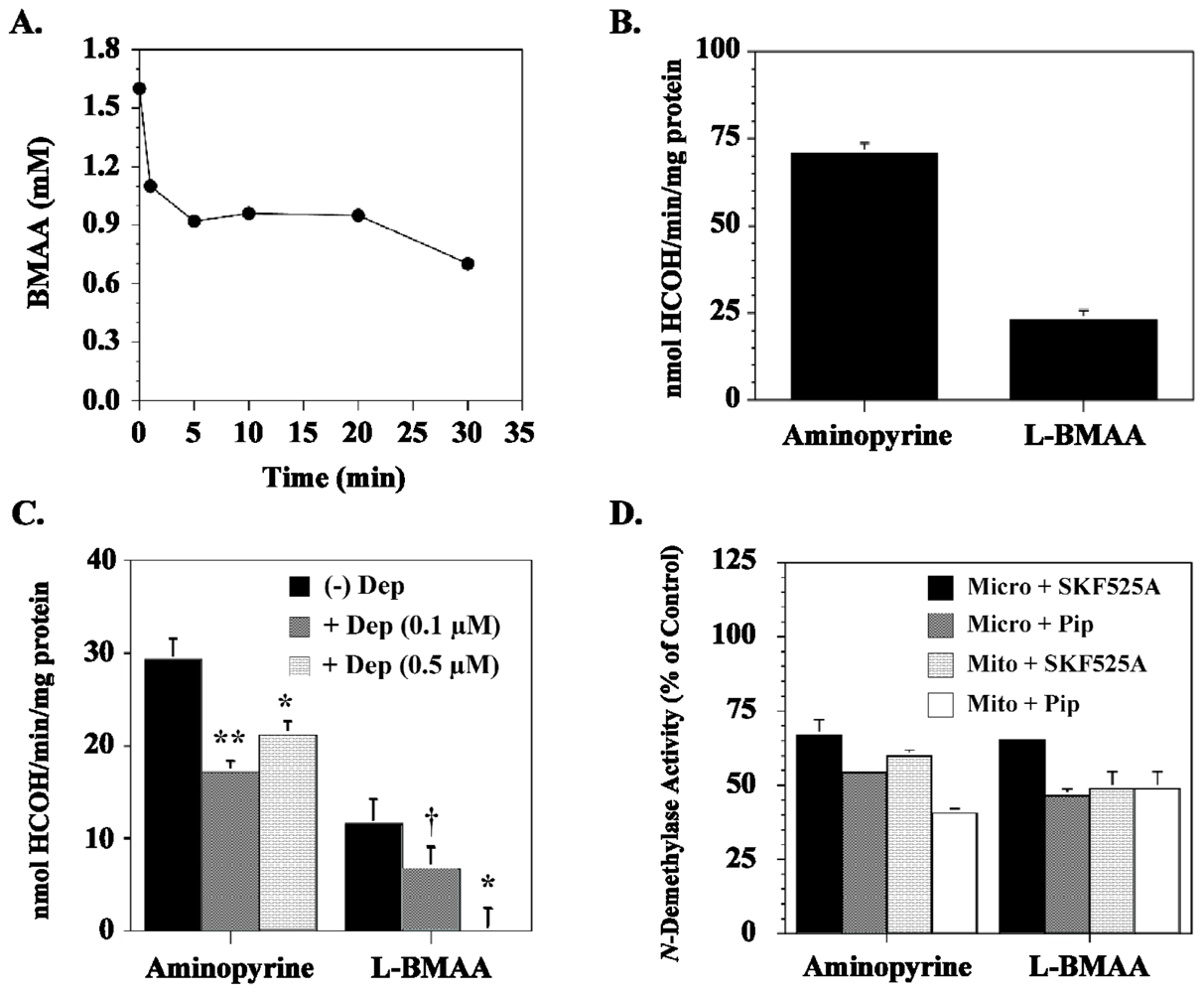
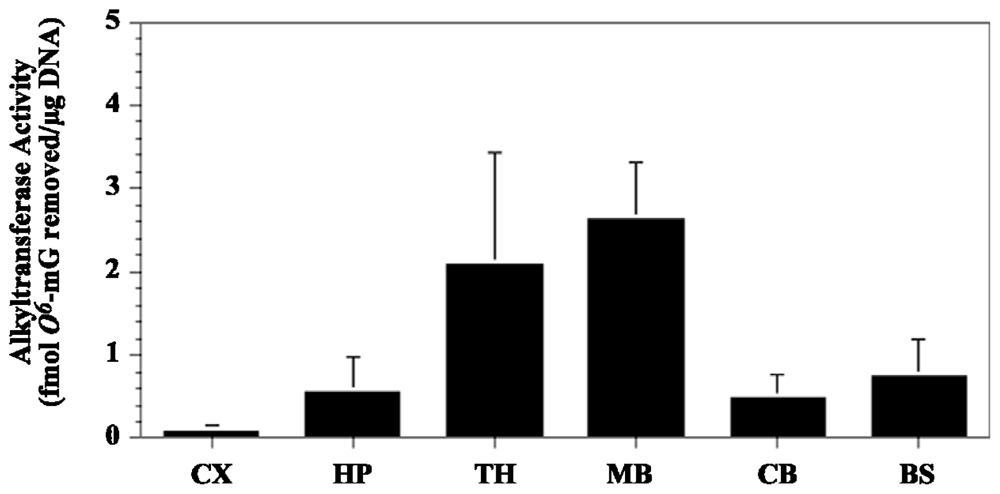
5. Cycasin and the Young Adult Brain
6. Relevance to Neurodegenerative Disorders
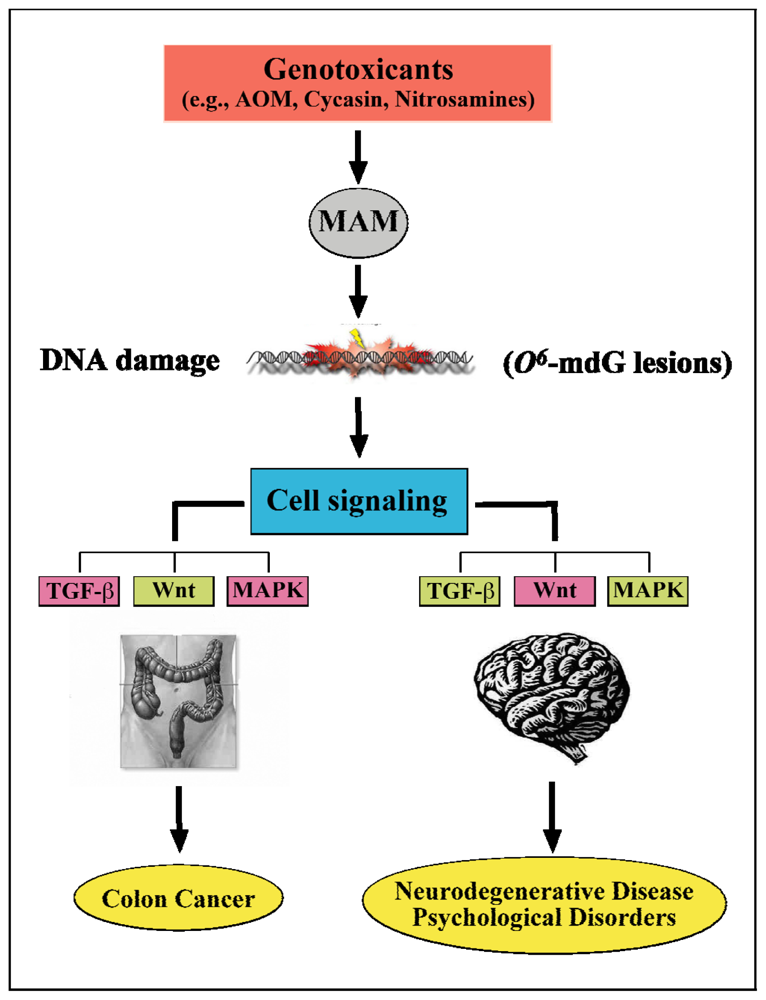
7. Summary and Putative Molecular Mechanisms
Acknowledgements
- Conflict of InterestThe authors declare no conflict of interest.
References and Notes
- Spencer, PS. Guam ALS/Parkinsonism-dementia: A long-latency neurotoxic disorder caused by “slow toxin(s)” in food? Can. J. Neurol. Sci 1987, 14, 347–357. [Google Scholar]
- Spencer, PS; Kisby, GE; Ludolph, AC. Slow toxins, biologic markers, and long-latency neurodegenerative disease in the western Pacific region. Neurology 1991, 41, 62–66. [Google Scholar]
- McGeer, PL; Steele, JC. The ALS/PDC syndrome of Guam: Potential biomarkers for an enigmatic disorder. 2011. [Google Scholar]
- Winton, MJ; Joyce, S; Zhukareva, V; Practico, D; Perl, DP; Galasko, D; Craig, U; Trojanowski, JQ; Lee, VM. Characterization of tau pathologies in gray and white matter of Guam parkinsonism-dementia complex. Acta Neuropathol 2006, 111, 401–412. [Google Scholar]
- Waring, SC; Esteban-Santillan, C; Reed, DM; Craig, UK; Labarthe, DR; Petersen, RC; Kurland, LT. Incidence of amyotrophic lateral sclerosis and of the parkinsonism-dementia complex of Guam, 1950–1989. Neuroepidemiology 2004, 23, 192–200. [Google Scholar]
- Kuzuhara, S; Kokubo, Y. Atypical parkinsonism of Japan: Amyotrophic lateral sclerosis-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease): An update. Mov. Disord 2005, 20, S108–S113. [Google Scholar]
- Mimuro, M; Kokubo, Y; Kuzuhara, S. Similar topographical distribution of neurofibrillary tangles in amyotrophic lateral sclerosis and parkinsonism-dementia complex in people living in the Kii peninsula of Japan suggests a single tauopathy. Acta Neuropathol 2007, 113, 653–658. [Google Scholar]
- Gajdusek, DC; Salazar, AM. Amyotrophic lateral sclerosis and parkinsonian syndromes in high incidence among the Auyu and Jakai people of West New Guinea. Neurology 1982, 32, 107–126. [Google Scholar]
- Spencer, PS; Palmer, VS; Ludolph, AC. On the decline and etiology of high-incidence motor system disease in West Papua (southwest New Guinea). Mov. Disord 2005, 20, 119–126. [Google Scholar]
- Gajdusek, DC; Zigas, V. Kuru. clinical, pathological and epidemiological study of an acute progressive degenerative disease of the central nervous system among natives of the Eastern Highlands of New Guinea. Am. J. Med 1959, 26, 442–469. [Google Scholar]
- Norrby, E. Prions and protein-folding diseases. J Intern Med 270 1–14.
- Galasko, D; Salmon, DP; Craig, UK; Thal, LJ; Schellenberg, G; Wiederholt, W. Clinical features and changing patterns of neurodegenerative disorders on Guam, 1997–2000. Neurology 2002, 58, 90–97. [Google Scholar]
- Sundar, PD; Yu, CE; Sieh, W; Steinbart, E; Garruto, RM; Oyanagi, K; Craig, U-K; Bird, TD; Wijsman, EM; Galasko, DR; Schellenberg, GD. Two sites in the MAPT region confer genetic risk for Guam ALS/PDC and dementia. Hum. Mol. Genet 2007, 16, 295–306. [Google Scholar]
- Tomiyama, H; Kokubo, Y; Sasaki, R; Li, Y; Imamichi, Y; Funayama, M; Mizuno, Y; Hattori, N; Kuzuhara, S. Mutation analyses in amyotrophic lateral sclerosis/parkinsonismdementia complex of the Kii peninsula, Japan. Mov Disord 2008, 23, 2344–2348. [Google Scholar]
- Hermosura, MC; Garruto, RM. TRPM7 and TRPM2-Candidate susceptibility genes for Western Pacific ALS and PD? Biochim. Biophys. Acta 2007, 1772, 822–835. [Google Scholar]
- Hermosura, MC; Nayakanti, H; Dorovkov, MV; Calderon, FR; Ryazanov, AG; Haymer, DS; Garruto, RM. A TRPM7 variant shows altered sensitivity to magnesium that may contribute to the pathogenesis of two Guamanian neurodegenerative disorders. Proc. Natl. Acad. Sci. USA 2005, 102, 11510–11515. [Google Scholar]
- Hara, K; Kokubo, Y; Ishiura, H; Fukuda, Y; Miyashita, A; Kuwano, R; Sasaki, R; Goto, J; Nishizawa, M; Kuzuhara, S; Tsuji, S. TRPM7 is not associated with amyotrophic lateral sclerosis-parkinsonism dementia complex in the Kii peninsula of Japan. Am. J. Med. Genet. B Neuropsychiatr. Genet 2010, 153B, 310–313. [Google Scholar]
- Kurland, LT. An appraisal of the neurotoxicity of cycad and the etiology of amyotrophic lateral sclerosis on Guam. Fed. Proc 1972, 31, 1540–1542. [Google Scholar]
- Oyanagi, K; Kawakami, E; Kikuchi-Horie, K; Ohara, K; Ogata, K; Takahama, S; Wada, M; Kihira, T; Yasui, M. Magnesium deficiency over generations in rats with special references to the pathogenesis of the Parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Neuropathology 2006, 26, 115–128. [Google Scholar]
- Ahlskog, JE; Waring, SC; Kurland, LT; Petersen, RC; Moyer, TP; Harmsen, WS; Maraganore, DM; O’Brien, PC; Esteban-Santillan, C; Bush, V. Guamanian neurodegenerative disease: Investigation of the calcium metabolism/heavy metal hypothesis. Neurology 1995, 45, 1340–1344. [Google Scholar]
- Whiting, M. Identification of toxic elements of cycads. In Toxicity of Cycads; Implications for Neurodegenerative Diseases and Cancer, Transcripts of the Four Cycad Conferences; Third World Medical Research Foundation, 1988; New York, NY, USA. [Google Scholar]
- Bell, EA; Vega, A; Nunn, PB. A neurotoxic amino acid in seeds of cycas circinalis. In Toxicity of Cycads; Implications for Neurodegenerative Diseases and Cancer, Transcripts of the Four Cycad Conferences; Third World Medical Research Foundation, 1988; New York, NY, USA. [Google Scholar]
- Spencer, PS; Nunn, PB; Hugon, J; Ludolph, AC; Ross, SM; Roy, DN; Robertson, RC. Guam amyotrophic lateral sclerosis-parkinsonism-dementia linked to a plant excitant neurotoxin. Science 1987, 237, 517–522. [Google Scholar]
- Banack, SA; Cox, PA. Biomagnification of cycad neurotoxins in flying foxes: Implications for ALS-PDC in Guam. Neurology 2003, 61, 387–389. [Google Scholar]
- Spencer, PS; Palmer, V; Herman, A; Asmedi, A. Cycad use and motor neuron disease in Irian Jaya. Lancet 1987, 2, 1273–1274. [Google Scholar]
- Spencer, PS; Ohta, M; Palmer, V. Cycad use and motor neuron disease in Kii peninsula of Japan. Lancet 1987, 2, 1462–1463. [Google Scholar]
- Kobayashi, A. Cycasin in cycad materials used in Japan. Fed. Proc 1972, 31, 1476–1477. [Google Scholar]
- Kisby, GE; Ellison, M; Spencer, PS. Content of the neurotoxins cycasin (methylazoxymethanol β-D-glucoside) and BMAA (β-N-methylamino-L-alanine) in cycad flour prepared by Guam Chamorros. Neurology 1992, 42, 1336–1340. [Google Scholar]
- Zegura, B; Straser, A; Filipic, M. Genotoxicity and potential carcinogenicity of cyanobacterial toxins - a review. Mutat. Res 2011, 727, 16–41. [Google Scholar]
- Ross, SM; Seelig, M; Spencer, PS. Specific antagonism of excitotoxic action of ‘uncommon’ amino acids assayed in organotypic mouse cortical cultures. Brain Res 1987, 425, 120–127. [Google Scholar]
- Allen, CN; Omelchenko, I; Ross, SM; Spencer, P. The neurotoxin, beta-N-methylamino-Lalanine (BMAA) interacts with the strychnine-insensitive glycine modulatory site of the N-methyl-D-aspartate receptor. Neuropharmacology 1995, 34, 651–658. [Google Scholar]
- Weiss, JH; Koh, J-Y; Choi, DW. Neurotoxicity of β-N-methylamino-L-alanine (BMAA) and β-N-oxalylamino-L-alanine (BOAA) on cultured cortical neurons. Brain Res 1989, 497, 64–71. [Google Scholar]
- Weiss, JH; Choi, DW. β-N-Methylamino-L-alanine neurotoxicity: Requirement for bicarbonate as a cofactor. Science 1994, 19, 973–975. [Google Scholar]
- Ross, SM; Spencer, PS. Specific antagonism of behavioral action of “uncommon” amino acids linked to motor-system diseases. Synapse 1987, 1, 248–253. [Google Scholar]
- Spencer, PS; Hugon, J; Ludolph, A; Nunn, PB; Ross, SM; Roy, DN; Schaumburg, HH. Discovery and partial characterization of primate motor-system toxins. Ciba Found. Symp 1987, 126, 221–238. [Google Scholar]
- Costa, LG; Giordano, G; Faustman, EM. Domoic acid as a developmental neurotoxin. Neurotoxicology 2010, 31, 409–423. [Google Scholar]
- Teitelbaum, JS; Zatorre, RJ; Carpenter, S; Gendron, D; Evans, AC; Gjedde, A; Cashman, NR. Neurologic sequelae of domoic acid intoxication due to the ingestion of contaminated mussels. N. Engl. J. Med 1990, 322, 1781–1787. [Google Scholar]
- Banack, SA; Murch, SJ; Cox, PA. Neurotoxic flying foxes as dietary items for the Chamorro people, Marianas Islands. J. Ethnopharmacol 2006, 106, 97–104. [Google Scholar]
- Murch, SJ; Cox, PA; Banack, SA; Steele, JC; Sacks, OW. Occurrence of beta-methylaminol- alanine (BMAA) in ALS/PDC patients from Guam. Acta Neurol. Scand 2004, 110, 267–269. [Google Scholar]
- Kisby, GE; Ross, SM; Spencer, PS; Gold, BG; Nunn, PB; Roy, DN. Cycasin and BMAA: Candidate neurotoxins for western Pacific amyotrophic lateral sclerosis/Parkinsonism-dementia complex. Neurodegeneration 1992, 1, 73–82. [Google Scholar]
- Spencer, PS; Ross, SM; Kisby, G; Roy, DN. Western Pacific amyotrophic lateral sclerosis: putative role of cycad toxins. In Amyotrophic Lateral Sclerosis: Concepts in Pathogenesis and Etiology; Hudson, AJ, Ed.; University of Toronto Press: Toronto, ON, Canada, 1990; pp. 263–295. [Google Scholar]
- Nunn, PB; Ponnusamy, M. Beta-N-methylaminoalanine (BMAA): Metabolism and metabolic effects in model systems and in neural and other tissues of the rat in vitro. Toxicon 2009, 54, 85–94. [Google Scholar]
- Kisby, GE; Nottingham, V; Kayton, R; Roy, DN; Spencer, PS. Brain metabolism of β-N-methylamino-L-alanine (BMAA) and protection of excitotoxicity by GABA-uptake inhibitors. Soc. Neurosci 1992, 18, 82. [Google Scholar]
- Ravindranath, V; Ananda Theertha Varada, HK. High activity of cytochrome P-450-linked aminopyrine N-demethylase in mouse brain microsomes, and associated sex-related difference. Biochem. J 1989, 261, 769–773. [Google Scholar]
- Kisby, GE; Roy, DN; Spencer, PS. Determination of β-N-methylamino-L-alanine (BMAA) in plant (Cycas circinalis L.) an animal tissue by precolumn derivatization with 9-fluorenylmethyl chloroformate (FMOC) and reversed-phase high-performance liquid chromotography. J. Neurosci. Methods 1988, 26, 45–54. [Google Scholar]
- Pai, KS; Ravindranath, V. Protection and potentiation of MPTP-induced toxicity by cytochrome P-450 inhibitors and inducer: In vitro studies with brain slices. Brain Res 1991, 555, 239–244. [Google Scholar]
- Pickard, MR; Sinha, AK; Gullo, D; Patel, N; Hubank, M; Ekins, RP. The effect of 3,5,3′-triiodothyronine on leucine uptake and incorporation into protein in cultured neurons and subcellular fractions of rat central nervous system. Endocrinology 1987, 121, 2018–2026. [Google Scholar]
- Kisby, GE; Kabel, H; Hugon, J; Spencer, P. Damage and repair of nerve cell DNA in toxic stress. Drug Metab. Rev 1999, 31, 589–618. [Google Scholar]
- Zhang, ZX; Anderson, DW; Mantel, N; Román, GC. Motor neuron disease on Guam: Geographic and familial occurrence, 1956–85. Acta Neurol. Scand 1996, 94, 51–59. [Google Scholar]
- Roman, GC; Zhang, Z-X; Ellenberg, JA. The Neuroepidemiology of Parkinson’s Disease; Marcel Dekker, 1995; New York, NY, USA. [Google Scholar]
- Spencer, PS; Dastur, DK. Neurolathyrism and neurocycadism. In Neurological Sciences—An Overview of Current Problems Section VI Tropical Neurology and NeurotoxicologyI; Dastur, DK, Shahani, M, Bharucha, EP, Eds.; Interprint: New Delhi, India, 1986; Volume Section VI. [Google Scholar]
- Spencer, PS; Kisby, GE; Palmer, VS; Obendorf, P. Cycasin, Methylazoxymethanol, and related compounds. In Experimental and Clinical Neurotoxicology, 2nd ed; Spencer, PS, Schaumburg, HH, Eds.; Oxford University Press: New York NY, USA, 1999; pp. 436–447. [Google Scholar]
- Sieber, SM; Correa, P; Dalgard, DW; McIntire, KR; Adamson, RH. Carcinogenicity and hepatotoxicity of cycasin and its aglycone methylazoxymethanol acetate in nonhuman primates. J. Natl. Cancer Inst 1980, 65, 177–189. [Google Scholar]
- Adamson, RH. Induction of hepatocellular carcinoma in nonhuman primates by chemical carcinogens. Cancer Detect. Prev 1989, 14, 215–219. [Google Scholar]
- Seawright, AA; Brown, AW; Nolan, CC; Cavanagh, JB. Selective degeneration of cerebellar cortical neurons caused by cycad neurotoxin, L-β-methylaminoalanine (L-BMAA), in rats. Neuropathol. Appl. Neurobiol 1990, 16, 153–169. [Google Scholar]
- Shen, WB; McDowell, KA; Siebert, AA; Clark, SM; Dugger, NV; Valentino, KM; Jinnah, HA; Sztalryd, C; Fishman, PS; Shaw, CA; Jafri, MS; Yarowsky, PJ. Environmental neurotoxin-induced progressive model of parkinsonism in rats. Ann. Neurol 2010, 68, 70–80. [Google Scholar]
- Spencer, PS; Lasarev, MR; Palmer, VS; Kisby, GE. Neurotoxic cycad components and Western Pacific ALS/PDC. Ann Neurol 2010, 68, 975–976, author reply 976. [Google Scholar]
- Caparros-Lefebvre, D; Elbaz, A. Possible relation of atypical parkinsonism in the French West Indies with consumption of tropical plants: A case-control study. Caribbean Parkinsonism Study Group. Lancet 1999, 354, 281–286. [Google Scholar]
- Lannuzel, A; Hoglinger, GU; Champy, P; Michel, PP; Hirsch, EC; Ruberg, M. Is atypical parkinsonism in the Caribbean caused by the consumption of Annonacae. J Neural Transm Suppl 2006, 60, 153–157. [Google Scholar]
- Spencer, PS; Ludolph, AC; Kisby, GE. Neurologic diseases associated with use of plant components with toxic potential. Environ.Res 1993, 62, 106–113. [Google Scholar]
- Laqueur, GL. Carcinogenic effects of cycad meal and cycasin, methylazoxymethanol glycoside, in rats and effects of cycasin in germfree rats. Fed. Proc 1964, 23, 1386–1388. [Google Scholar]
- Moore, MA; Baumann, F; Foliaki, S; Goodman, MT; Haddock, R; Maraka, R; Koroivueta, J; Roder, D; Vinit, T; Whippy, HJ; Sobue, T. Cancer epidemiology in the Pacific islands—past, present and future. Asian Pac. J. Cancer Prev 2010, S2, 99–106. [Google Scholar]
- Borenstein, AR; Mortimer, JA; Schofield, E; Wu, Y; Salmon, DP; Gamst, A; Olichney, J; Thal, LJ; Silbert, L; Kaye, J; Craig, UL; Schellenberg, GD; Galasko, DR. Cycad exposure and risk of dementia, MCI, and PDC in the Chamorro population of Guam. Neurology 2007, 68, 1764–1771. [Google Scholar]
- Shiraki, H; Yase, Y. Amyotrophic Lateral Sclerosis in Japan. In Handbook of Clinical Neurology Vol 22 System Disorders and Atrophy, Part 2; Vinken, PJ, Bruyn, GW, Eds.; American Elsevier: New York, NY, USA, 1975; pp. 353–419. [Google Scholar]
- Husseman, JW; Nochlin, D; Vincent, I. Mitotic activation: A convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol. Aging 2000, 21, 815–828. [Google Scholar]
- Sullivan-Jones, P; Gouch, AB; Holson, RR. Postnatal methylazoxymethanol: Sensitive periods and regional selectivity of effects. Neurotoxicol. Teratol 1994, 16, 631–637. [Google Scholar]
- Vincent, I; Pae, CI; Hallows, JL. The cell cycle and human neurodegenerative disease. Prog. Cell Cycle Res 2003, 5, 31–41. [Google Scholar]
- McShea, A; Lee, HG; Petersen, RB; Casadesus, G; Vincent, I; Linford, NJ; Funk, JO; Shapiro, RA; Smith, MA. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim. Biophys. Acta 2007, 1772, 467–472. [Google Scholar]
- Zhu, X; Siedlak, SL; Wang, Y; Perry, G; Castellani, RJ; Cohen, ML; Smith, MA. Neuronal binucleation in Alzheimer disease hippocampus. Neuropathol. Appl. Neurobiol 2008, 34, 457–465. [Google Scholar]
- Landrigan, PJ; Sonawane, B; Butler, RN; Trasande, L; Callan, R; Droller, D. Early environmental origins of neurodegenerative disease in later life. Environ. Health Perspect 2005, 113, 1230–1233. [Google Scholar]
- Plun-Favreau, H; Lewis, PA; Hardy, J; Martins, LM; Wood, NW. Cancer and neurodegeneration: Between the devil and the deep blue sea. PLoS Genet 2010, 6, e1001257. [Google Scholar]
- Morris, LG; Veeriah, S; Chan, TA. Genetic determinants at the interface of cancer and neurodegenerative disease. Oncogene 2010, 29, 3453–3464. [Google Scholar]
- Nagata, Y; Matsumoto, H. Studies on methylazoxymethanol: Methylation of nucleic acids in the fetal rat brain. Proc. Soc. Exp. Biol. Med 1969, 132, 383–385. [Google Scholar]
- Matsumoto, H; Spatz, M; Laqueur, GL. Quantitative changes with age in the DNA content of methylazoxymethanol-induced microencephalic rat brain. J. Neurochem 1972, 19, 297–306. [Google Scholar]
- Kisby, GE; Standley, M; Lu, X; O’Malley, J; Lin, B; Muniz, J; Luo, NL; Pattee, P; Back, SA; Nagalla, SR. Molecular networks perturbed in a developmental animal model of brain injury. Neurobiol. Dis 2005, 19, 108–118. [Google Scholar]
- Kisby, GE; Olivas, A; Park, T; Churchwell, M; Doerge, D; Samson, LD; Gerson, SL; Turker, MS. DNA repair modulates the vulnerability of the developing brain to alkylating agents. DNA Repair 2009, 8, 400–412. [Google Scholar] [Green Version]
- Samson, LD. The repair of DNA alkylation damage by methyltransferases and glycosylases. Essays Biochem 1992, 27, 69–78. [Google Scholar]
- Kleihues, P; Bucheler, J. Long-term persistence of O6-methylguanine in rat brain DNA. Nature 1977, 269, 625–626. [Google Scholar]
- Kreklau, EL; Liu, N; Li, Z; Cornetta, K; Erickson, LC. Comparison of single- versus double-bolus treatments of O(6)-benzylguanine for depletion of O(6)-methylguanine DNA methyltransferase (MGMT) activity in vivo: Development of a novel fluorometric oligonucleotide assay for measurement of MGMT activity. J. Pharmacol. Exp. Ther 2001, 297, 524–530. [Google Scholar]
- Burns, JA; Dreij, K; Cartularo, L; Scicchitano, DA. O6-Methylguanine induces altered proteins at the level of transcription in human cells. Nucleic Acids Res 2010, 38, 8178–8187. [Google Scholar]
- Silber, JR; Blank, A; Bobola, MS; Mueller, BA; Kolstoe, DD; Ojemann, GA; Berger, MS. Lack of the DNA repair protein O6-methylguanine-DNA methyltransferase in histologically normal brain adjacent to primary human brain tumors. Proc. Natl. Acad. Sci. USA 1996, 93, 6941–6946. [Google Scholar]
- Bobola, MS; Blank, A; Berger, MS; Silber, JR. O6-Methylguanine-DNA methyltransferase deficiency in developing brain: Implications for brain tumorigenesis. DNA Repair (Amst.) 2007, 6, 1127–1133. [Google Scholar]
- Kisby, GE; Standley, M; Park, T; Olivas, A; Fei, S; Jacob, T; Reddy, A; Lu, X; Pattee, P; Nagalla, SR. Proteomic analysis of the genotoxicant methylazoxymethanol (MAM) induced changes in the developing cerebellum. J. Proteome Res 2006, 5, 2656–2665. [Google Scholar]
- Kisby, GE; Olivas, A; Standley, M; Lu, X; Pattee, P; O’Malley, J; Li, X; Muniz, J; Nagalla, R. Genotoxicants target distinct molecular networks in neonatal neurons. Environ. Health. Perspect 2006, 114, 1703–1712. [Google Scholar]
- Yang, W; Woltjer, RL; Sokal, I; Pan, C; Wang, Y; Brodey, M; Peskind, ER; Leverenz, JB; Zhang, J; Perl, DP; Galasko, DR; Montine, TJ. Quantitative proteomics identifies surfactant-resistant alpha-synuclein in cerebral cortex of parkinsonism-dementia complex of Guam but not Alzheimer’s disease or progressive supranuclear palsy. Am. J. Pathol 2007, 171, 993–1002. [Google Scholar]
- Coppede, F; Migliore, L. DNA damage and repair in Alzheimer’s disease. Curr. Alzheimer Res 2009, 6, 36–47. [Google Scholar]
- Lovell, MA; Markesbery, WR. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res 2007, 35, 7497–7504. [Google Scholar]
- Martin, LJ. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol 2008, 67, 377–387. [Google Scholar]
- Nakabeppu, Y; Tsuchimoto, D; Yamaguchi, H; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res 2007, 85, 919–934. [Google Scholar]
- Kisby, GE; Fry, RC; Lasarev, MR; Bammler, TK; Beyer, RP; Churchwell, M; Doerge, DR; Meira, LB; Palmer, VS; Ramos-Crawford, AL; et al. The cycad genotoxin MAM modulates brain cellular pathways involved in neurodegenerative disease and cancer in a DNA damagelinked manner. PLoS One 2011, 6, e20911. [Google Scholar]
- Buecheler, J; Kleihues, P. Excision of O6-methylguanine from DNA of various mouse tissues following a single injection of N-methyl-nitrosourea. Chem. Biol. Interact 1977, 16, 325–333. [Google Scholar]
- Likhachev, AJ; Alekandrov, VA; Anisimov, VN; Bespalov, VG; Korsakov, MV; Ovsyannikov, AI; Popovich, IG; Napalkov, NP; Tomatis, L. Persistence of methylated purines in the DNA of various rat fetal and maternal tissues and carcinogenesis in the offspring following a single transplacental dose of N-methyl-N-nitrosourea. Int. J. Cancer 1983, 31, 779–784. [Google Scholar]
- Balduini, W; Cimino, M; Lombardelli, G; Abbracchio, MP; Peruzzi, G; Cecchini, T; Gazzanelli, GC; Cattabeni, F. Microencephalic rats as a model for cognitive disorders. Clin. Neuropharmacol 1986, 9, S8–S18. [Google Scholar]
- Cattabeni, F; Di Luca, M. Developmental models of brain dysfunctions induced by targeted cellular ablations with methylazoxymethanol. Physiol. Rev 1997, 77, 199–215. [Google Scholar]
- Laqueur, GL. Carcinogenic effects of cycad meal and cycasin, methylazoxymethanol glycoside, in rats and effects of cycasin in germfree rats. Fed. Proc 1964, 23, 1386–1388. [Google Scholar]
- Nigro, ND. Animal model for colorectal cancer. Prog. Clin. Biol. Res 1985, 186, 161–173. [Google Scholar]
- Garruto, RM; Gadjusek, DC; Chen, K-M. Amyotrophic lateral sclerosis among Chamorro migrants from Guam. Ann. Neurol 1980, 8, 612–619. [Google Scholar]
- Garruto, RM; Gadjusek, DC; Chen, K-M. Amyotrophic lateral sclerosis and parkinsonism-dementia among Filipino migrants to Guam. Ann. Neurol 1981, 10, 341–350. [Google Scholar]
- Anderson, FH; Richardson, EP; Okazaki, H; Brody, JA. Neurofibrillary degeneration on Guam: Frequency in Chamorros and non Chamorros with no known neurological disease. Brain 1979, 102, 65–77. [Google Scholar]
- Kisby, GE; Palmer, VS; Lasarev, MR; Fry, R; Iordanov, M; Magun, E; Samson, L; Spencer, P. Does the cycad genotoxin MAM implicated in Guam ALS-PDC induce disease-relevant changes in mouse brain that includes olfaction? Commun Integr Biol.
- Doty, RL; Perl, DP; Steele, JC; Chen, KM; Pierce, JD, Jr; Reyes, P; Kurland, LT. Odor identification deficit of the Parkinsonism-dementia complex of Guam: Equivalence to that of Alzheimer's and idiopathic Parkinson’s disease. Neurology 1991, 41, 77–80. [Google Scholar]
- Ahlskog, JE; Waring, SC; Petersen, RC; Esteban-Santillan, C; Craig, UK; O’Brien, PC; Plevak, MF; Kurland, LT. Olfactory dysfunction in Guamanian ALS, parkinsonism, and dementia. Neurology 1998, 51, 1672–1677. [Google Scholar]
- Wilson, RS; Arnold, SE; Schneider, JA; Boyle, PA; Buchman, AS; Bennett, DA. Olfactory impairment in presymptomatic Alzheimer’s disease. Ann. N. Y. Acad. Sci 2009, 1170, 730–735. [Google Scholar]
- Berendse, HW; Roos, DS; Raijmakers, P; Doty, RL. Motor and non-motor correlates of olfactory dysfunction in Parkinson’s disease. J Neurol Sci. [CrossRef]
- Doty, RL; Perl, DP; Steele, JC; Chen, KM; Pierce, JD, Jr; Reyes, P; Kurland, LT. Olfactory dysfunction in three neurodegenerative diseases. Geriatrics 1991, 46S, 47–51. [Google Scholar]
- Staropoli, JF. Tumorigenesis and neurodegeneration: Two sides of the same coin? Bioessays 2008, 30, 719–727. [Google Scholar]
- Caricasole, A; Bakker, A; Copani, A; Nicoletti, F; Gaviraghi, G; Terstappen, GC. Two sides of the same coin: Wnt signaling in neurodegeneration and neuro-oncology. Biosci. Rep 2005, 25, 309–327. [Google Scholar]
- Muyllaert, D; Kremer, A; Jaworski, T; Borghgraef, P; Devijver, H; Croes, S; Dewachter, I; van Leuven, F. Glycogen synthase kinase-3beta, or a link between amyloid and tau pathology? Genes Brain Behav 2008, 7S, 57–66. [Google Scholar]
- Behrens, MI; Lendon, C; Roe, CM. A common biological mechanism in cancer and Alzheimer’s disease? Curr. Alzheimer Res 2009, 6, 196–204. [Google Scholar]
- Souter, S; Lee, G. Tubulin-independent tau in Alzheimer’s disease and cancer: Implications for disease pathogenesis and treatment. Curr. Alzheimer Res 2010, 7, 697–707. [Google Scholar]
- Serrano, J; Fernandez, AP; Martinez-Murillo, R; Martinez, A. High sensitivity to carcinogens in the brain of a mouse model of Alzheimer’s disease. Oncogene 2010, 29, 2165–2171. [Google Scholar]
- Chen, J; Huang, XF. The signal pathways in azoxymethane-induced colon cancer and preventive implications. Cancer Biol. Ther 2009, 8, 1313–1317. [Google Scholar]
- Wali, RK; Skarosi, S; Hart, J; Zhang, Y; Dolan, ME; Moschel, RC; Nguyen, L; Mustafi, R; Brasitus, TA; Bissonnette, M. Inhibition of O(6)-methylguanine-DNA methyltransferase increases azoxymethane-induced colonic tumors in rats. Carcinogenesis 1999, 20, 2355–2360. [Google Scholar]
- Takahashi, M; Nakatsugi, S; Sugimura, T; Wakabayashi, K. Frequent mutations of the beta-catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis 2000, 21, 1117–1120. [Google Scholar]
- Bonfanti, M; Broggini, M; Prontera, C; D’Incalci, M. O6-Methylguanine inhibits the binding of transcription factors to DNA. Nucleic Acids Res 1991, 19, 5739–5742. [Google Scholar]
- Ghosh, R; Mitchell, DL. Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res 1999, 27, 3213–3218. [Google Scholar]
- Parsian, AJ; Funk, MC; Tao, TY; Hunt, CR. The effect of DNA damage on the formation of protein/DNA complexes. Mutat. Res 2002, 501, 105–113. [Google Scholar]
- Hailer-Morrison, MK; Kotler, JM; Martin, BD; Sugden, KD. Oxidized guanine lesions as modulators of gene transcription. Altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2′-deoxyguanosine lesions in the NF-kappaB promoter element. Biochemistry 2003, 42, 9761–9770. [Google Scholar]
- Yamini, B; Yu, X; Dolan, ME; Wu, MH; Kufe, DW; Weichselbaum, RR. Inhibition of nuclear factor-kappaB activity by temozolomide involves O6-methylguanine induced inhibition of p65 DNA binding. Cancer Res 2007, 67, 6889–6898. [Google Scholar]
- Bregeon, D; Peignon, PA; Sarasin, A. Transcriptional mutagenesis induced by 8-oxoguanine in mammalian cells. PLoS Genet 2009, 5, e1000577. [Google Scholar]
- Bregeon, D; Doetsch, PW. Transcriptional mutagenesis: Causes and involvement in tumour development. Nat. Rev. Cancer 2011, 11, 218–227. [Google Scholar]
- de la Monte, SM; Neusner, A; Chu, J; Lawton, M. Epidemiological trends strongly suggest exposures as etiologic agents in the pathogenesis of sporadic Alzheimer’s disease, diabetes mellitus, and non-alcoholic steatohepatitis. J. Alzheimers Dis 2009, 17, 519–529. [Google Scholar]
- McCutcheon, JW. Nitrosamines in bacon: A case study of balancing risks. Public Health Rep 1984, 99, 360–364. [Google Scholar]
- Liu, C; Russell, RM. Nutrition and gastric cancer risk: An update. Nutr. Rev 2008, 66, 237–249. [Google Scholar]
- Lijinsky, W. N-Nitroso compounds in the diet. Mutat. Res 1999, 443, 129–138. [Google Scholar]
- Le Roux, J; Gallard, H; Croue, JP. Chloramination of nitrogenous contaminants (pharmaceuticals and pesticides): NDMA and halogenated DBPs formation. Water Res 2011, 45, 3164–3174. [Google Scholar]
- Oury, B; Limasset, JC; Protois, JC. Assessment of exposure to carcinogenic N-nitrosamines in the rubber industry. Int. Arch. Occup. Environ. Health 1997, 70, 261–271. [Google Scholar]
- Bolzan, AD; Bianchi, MS. Genotoxicity of streptozotocin. Mutat. Res 2002, 512, 121–134. [Google Scholar]
- Ahles, TA; Saykin, AJ. Candidate mechanisms for chemotherapy-induced cognitive changes. Nat. Rev. Cancer 2007, 7, 192–201. [Google Scholar]
- Wefel, JS; Kayl, AE; Meyers, CA. Neuropsychological dysfunction associated with cancer and cancer therapies: A conceptual review of an emerging target. Br. J. Cancer 2004, 90, 1691–1696. [Google Scholar]
- Thomson, MA; Green, CR; Rasma, BB; Holtshouser, JL; Hinderer, RK; Papciak, RJ; Leung, H; Howe, SR; Kuhn, TD. N-Nitrosamines in Patty’s Industrial Hygiene and Toxicology, 4th ed; Clayton, GD, Clayton, FE, Eds.; John Wiley & Sons: Etobicoke, ON, Canada, 1993; Volume Chapter 11, pp. 665–668. [Google Scholar]
- Salkovic-Petrisic, M; Hoyer, S. Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: An experimental approach. J. Neural Transm. Suppl 2007, 72, 217–233. [Google Scholar]
- de la Monte, SM; Tong, M. Mechanisms of nitrosamine-mediated neurodegeneration: Potential relevance to sporadic Alzheimer’s disease. J. Alzheimers Dis 2009, 17, 817–825. [Google Scholar]
- Agrawal, R; Tyagi, E; Shukla, R; Nath, C. A study of brain insulin receptors, AChE activity and oxidative stress in rat model of ICV STZ induced dementia. Neuropharmacology 2009, 56, 779–787. [Google Scholar]
- Clodfelder-Miller, BJ; Zmijewska, AA; Johnson, GV; Jope, RS. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 2006, 55, 3320–3325. [Google Scholar]
- Ke, YD; Delerue, F; Gladbach, A; Gotz, J; Ittner, LM. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One 2009, 4, e7917. [Google Scholar]
- Salkovic-Petrisic, M; Tribl, F; Schmidt, M; Hoyer, S; Riederer, P. Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J. Neurochem 2006, 96, 1005–1015. [Google Scholar]
- Tong, M; Longato, L; de la Monte, SM. Early limited nitrosamine exposures exacerbate high fat diet-mediated type 2 diabetes and neurodegeneration. BMC Endocr. Disord 2010, 10, 4. [Google Scholar]
- Kihira, T; Suzuki, A; Kondo, T; Wakayama, I; Yoshida, S; Hasegawa, K; Garruto, RM. Immunohistochemical expression of IGF-I and GSK in the spinal cord of Kii and Guamanian ALS patients. Neuropathology 2009, 29, 548–558. [Google Scholar]
- Craft, S. Insulin resistance and Alzheimer’s disease pathogenesis: Potential mechanisms and implications for treatment. Curr. Alzheimer Res 2007, 4, 147–152. [Google Scholar]
- Eikelenboom, P; van Exel, E; Hoozemans, JJ; Veerhuis, R; Rozemuller, AJ; van Gool, WA. Neuroinflammation - an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegen. Dis 2010, 7, 38–41. [Google Scholar]
- Gallo, V; Bueno-De-Mesquita, HB; Vermeulen, R; Andersen, PM; Kyrozis, A; Linseisen, J; Kaaks, R; Allen, NE; Roddam, AW; Boshuizen, HC; et al. Smoking and risk for amyotrophic lateral sclerosis: analysis of the EPIC cohort. Ann. Neurol 2009, 65, 378–385. [Google Scholar]
- Weisskopf, MG; Morozova, N; O’Reilly, EJ; McCullough, ML; Calle, EE; Thun, MJ; Ascherio, A. Prospective study of chemical exposures and amyotrophic lateral sclerosis. J Neurol Neurosur Ps 2009, 80, 558–561. [Google Scholar]
- Esclaire, F; Kisby, GE; Milne, J; Lesort, M; Spencer, P; Hugon, J. The Guam cycad toxin methylazoxymethanol damages neuronal DNA and modulates tau mRNA expression and excitotoxicity. Exp. Neurol 1999, 155, 11–21. [Google Scholar]
- Hugon, J; Esclaire, F; Lesort, M; Kisby, G; Spencer, P. Toxic neuronal apoptosis and modifications of tau and APP gene and protein expressions. Drug Metab. Rev 1999, 31, 635–647. [Google Scholar]
- Murch, SJ; Cox, PA; Banack, SA. A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc. Natl. Acad. Sci. USA 2004, 101, 12228–12231. [Google Scholar]
- Prusiner, SB; DeArmond, SJ. Molecular biology and pathology of scrapie and the prion diseases of humans. Brain Pathol 1991, 1, 297–310. [Google Scholar]
- Sikorska, B; Liberski, PP; Sobow, T; Budka, H; Ironside, JW. Ultrastructural study of florid plaques in variant Creutzfeldt-Jakob disease: A comparison with amyloid plaques in kuru, sporadic Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker disease. Neuropathol. Appl. Neurobiol 2009, 35, 46–59. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | N-Nitrosamine † | Concentration |
|---|---|---|
| Food/Beverages | ||
| Bacon | DMN, DEN, NPYR | 1–40 ppb |
| Salami | DMN | 10–80 ppb |
| Luncheon meat | DMN, DEN | 1–4 ppb |
| Hamburger | DMN | 15–25 ppb |
| Chinese marine salted fish | DMN DEN | 0.05–21 ppm |
| Smoked salmon | DMN | 0–26 ppb |
| Cheese | DMN | 1–4 ppb |
| Wheat flour | DEN | 0–10 ppb |
| Beer | DMA | |
| Commercial Products | ||
| Latex gloves | DMN | 37–329 ppb |
| DEN | < 10 μg/kg | |
| Cigarettes | DMN, DEN, NPYR | 0–180 ng/cig. |
| Pesticide Formulations | DMN | 300 ppb–640 ppm |
| Rubber nipples | DMN, DEN | < 100–200 μg/kg |
| Rubber toys | DMN | < 25 μg/kg |
| Rubber balloons | DMN | < 150 μg/kg |
| DEN | > 30 μg/kg | |
| Industrial Exposure | ||
| Leather Tanneries | DMN | 0.1–47 μg/m3 |
| Foundries, USA | DMN | 0.062–2.4 μg/m3 |
| Rubber, curing, salt bath | DMN, DEN | |
| Rubber, curing, injection molding | DMN | |
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kisby, G.E.; Spencer, P.S. Is Neurodegenerative Disease a Long-Latency Response to Early-Life Genotoxin Exposure? Int. J. Environ. Res. Public Health 2011, 8, 3889-3921. https://doi.org/10.3390/ijerph8103889
Kisby GE, Spencer PS. Is Neurodegenerative Disease a Long-Latency Response to Early-Life Genotoxin Exposure? International Journal of Environmental Research and Public Health. 2011; 8(10):3889-3921. https://doi.org/10.3390/ijerph8103889
Chicago/Turabian StyleKisby, Glen E., and Peter S. Spencer. 2011. "Is Neurodegenerative Disease a Long-Latency Response to Early-Life Genotoxin Exposure?" International Journal of Environmental Research and Public Health 8, no. 10: 3889-3921. https://doi.org/10.3390/ijerph8103889
APA StyleKisby, G. E., & Spencer, P. S. (2011). Is Neurodegenerative Disease a Long-Latency Response to Early-Life Genotoxin Exposure? International Journal of Environmental Research and Public Health, 8(10), 3889-3921. https://doi.org/10.3390/ijerph8103889