
Process Analytical Technology for the Production of Parenteral Lipid Emulsions According to Good Manufacturing Practices



Abstract
:1. Introduction
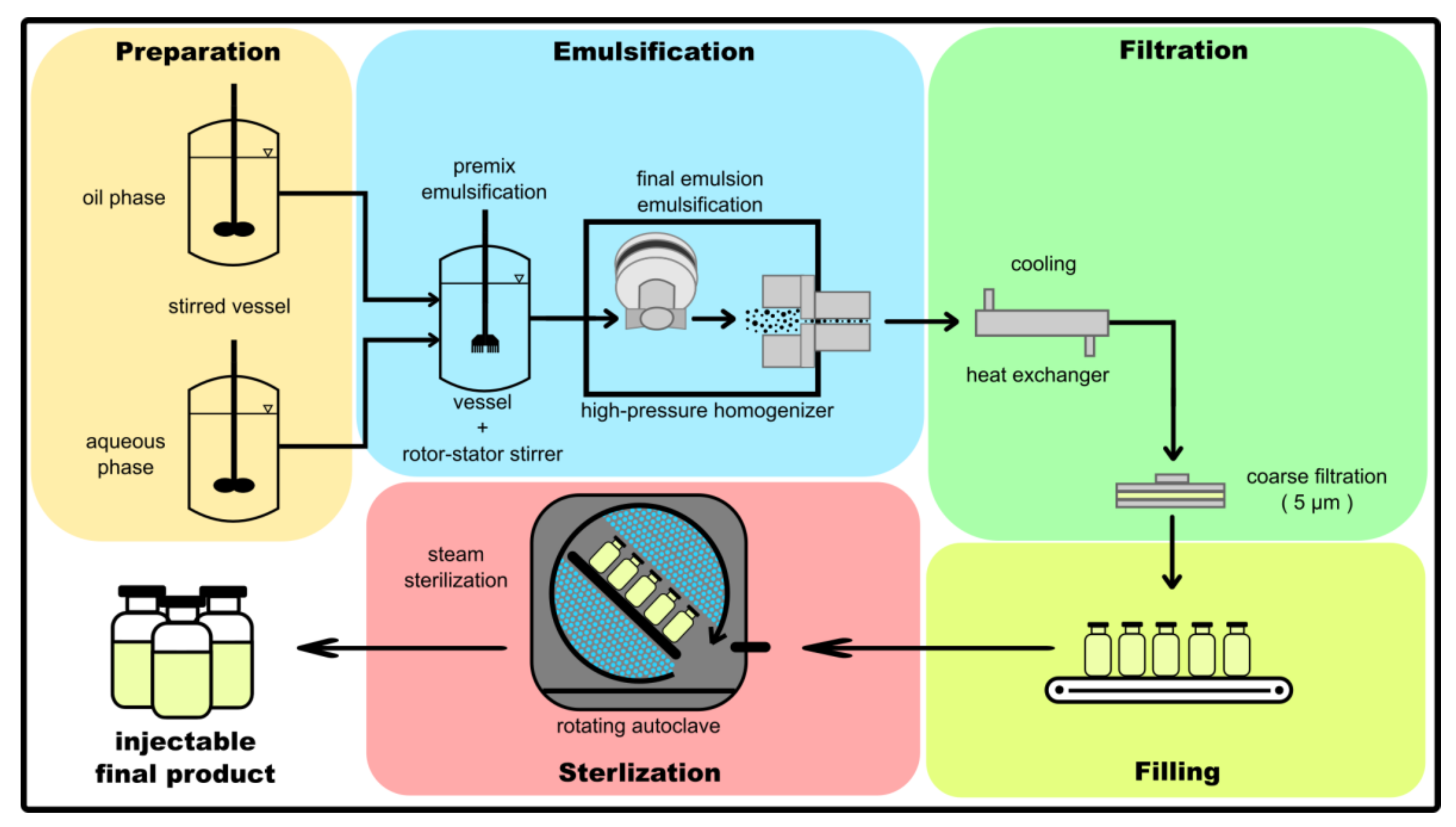
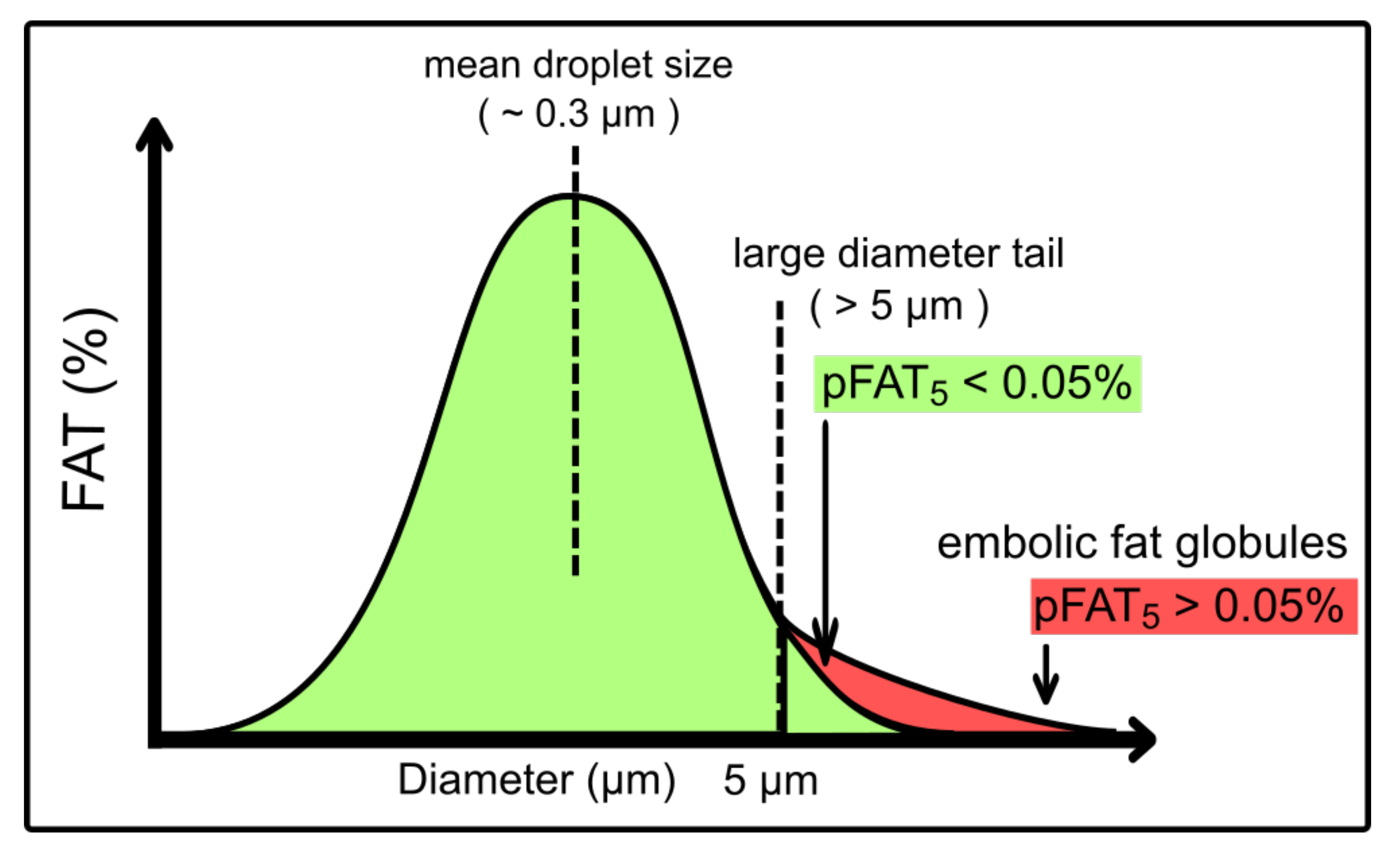
2. Considerations for the Manufacturing of Parenteral Lipid Emulsions
3. Regulatory Considerations
3.1. Process Validation and Quality by Design
3.2. Aseptic Processing of Parenteral Lipid Emulsions
4. PAT for Droplet Monitoring
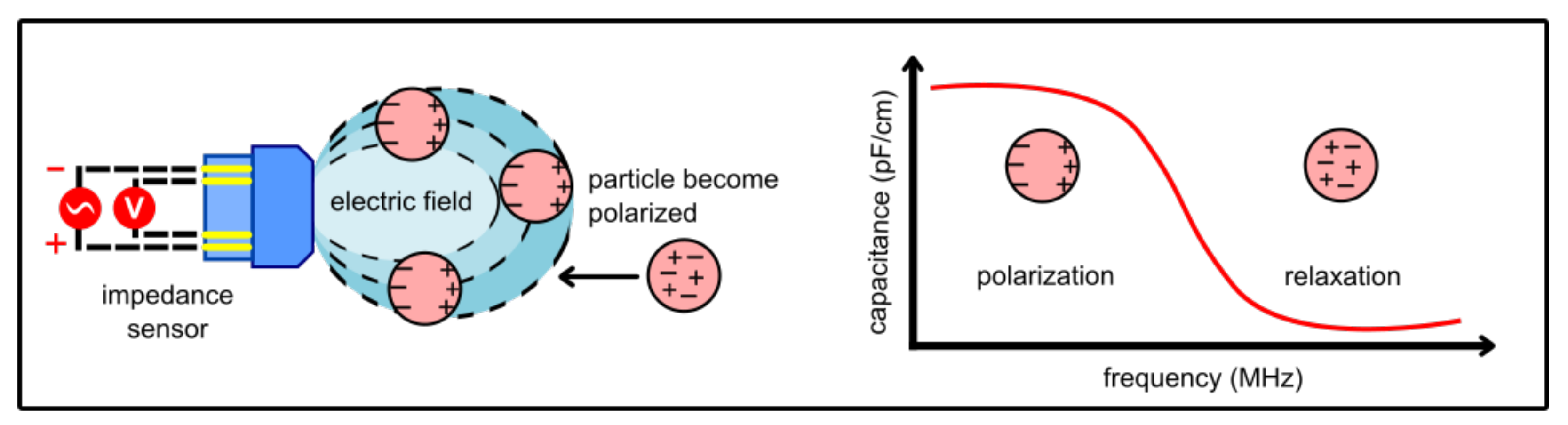
4.1. Dielectric Spectroscopy
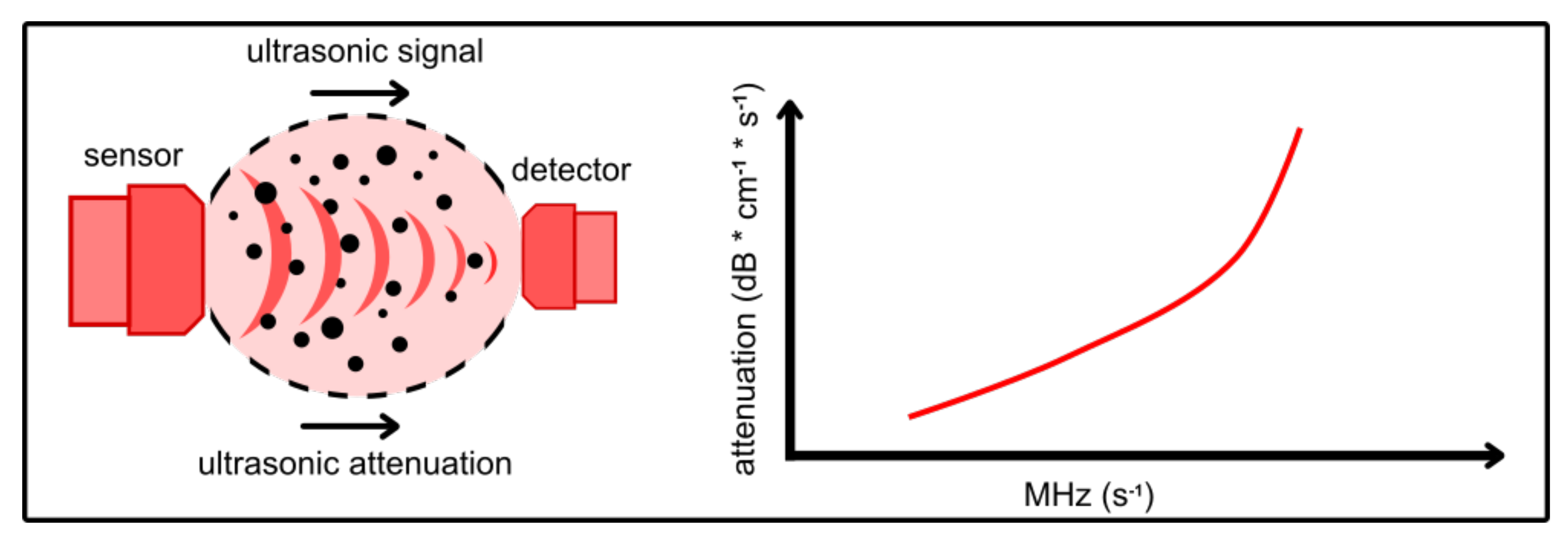
4.2. Ultrasonic Backscattering
4.3. Ultrasonic Attenuation Spectroscopy
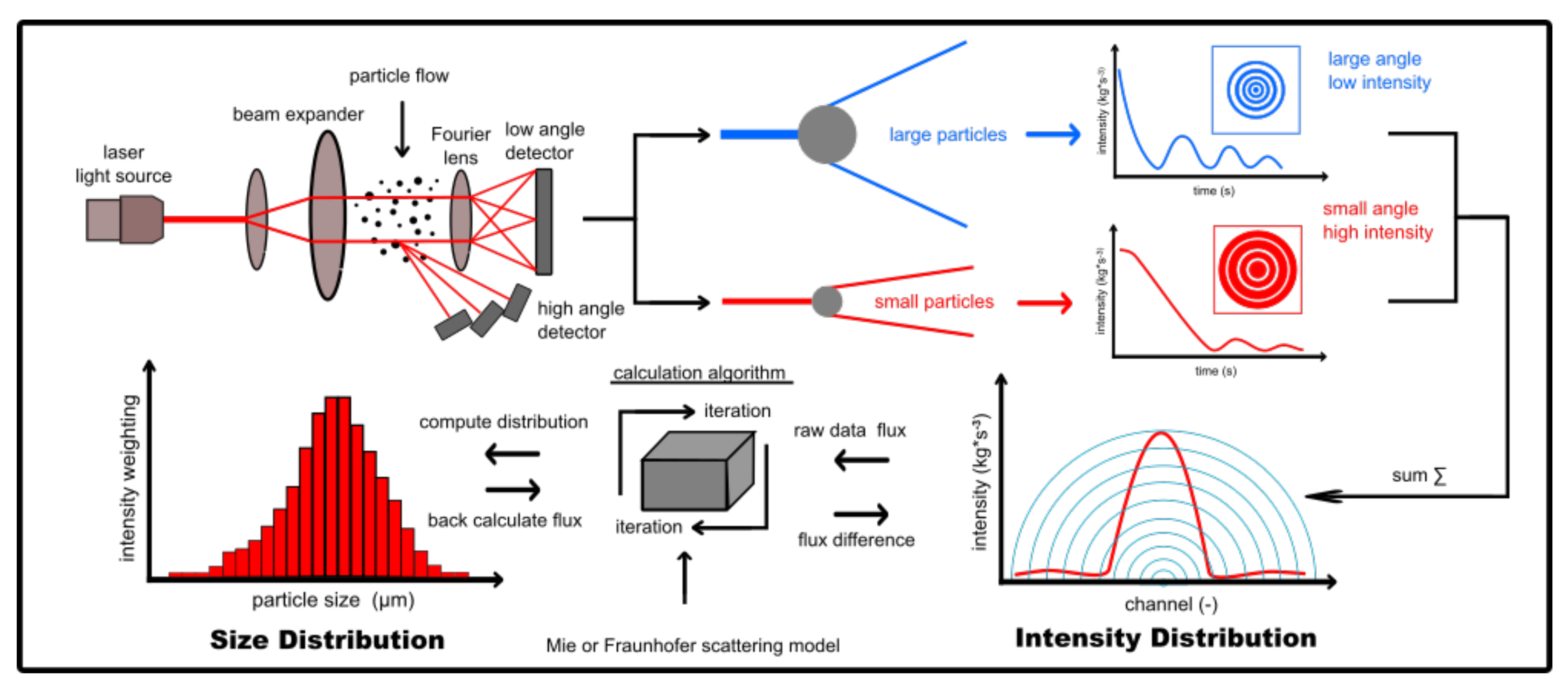
4.4. Laser Diffraction
4.5. Diffusing Wave Spectroscopy
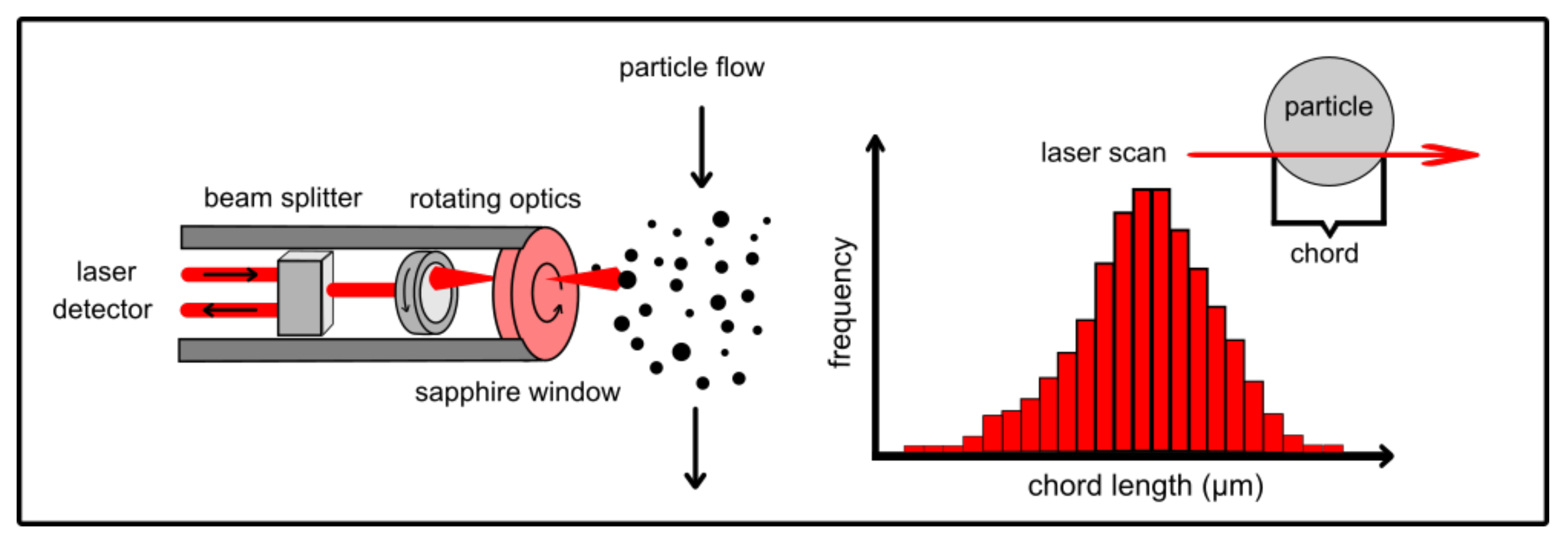
4.6. Laser Scanner Technology
4.7. Photo-Optical Sensors and Image Analysis
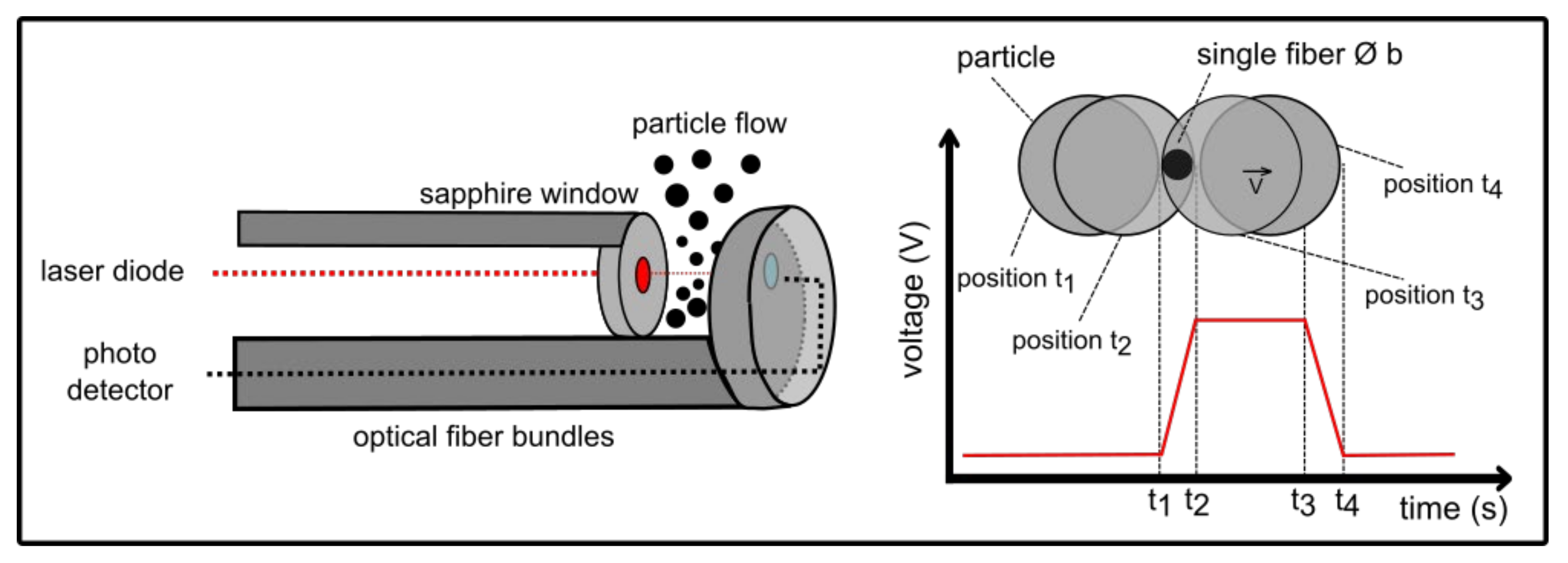
4.8. Spatial Filtering Technique
5. Conclusions
5.1. Considerations for the Manufacturing of Parenteral Lipid Emulsions
5.2. Regulatory Considerations for the Aseptic Processing of Parenteral Lipid Emulsions
5.3. PAT for Inline Droplet Monitoring
6. Outlook and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agalloco, J.; Akers, J.; Madesen, R. The Future of parenteral manufacturing. In Parenteral Medications, 4th ed.; CRC Press: Boca Raton, FL, USA, 2019; ISBN 9781498719148. [Google Scholar] [CrossRef]
- Cardona, D.; Nadal, M.; Estelrich, J.; Mangues, M.A. Review of drug stability in parenteral nutrition admixtures. e-SPEN J. 2013, 8, e135–e140. [Google Scholar] [CrossRef]
- Driscoll, D.F. Pharmaceutical and Clinical Aspects of Lipid Injectable Emulsions. J. Parenter. Enter. Nutr. 2016, 41, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Rosi Cappellani, M.; Perinelli, D.R.; Pescosolido, L.; Schoubben, A.; Cespi, M.; Cossi, R.; Blasi, P. Injectable nanoemulsions prepared by high pressure homogenization: Processing, sterilization, and size evolution. Appl. Nanosci. 2018, 8, 1483–1491. [Google Scholar] [CrossRef]
- Boullata, J.I.; Mirtallo, J.M.; Sacks, G.S.; Salman, G.; Gura, K.; Canada, T.; Maguire, A. The ASPEN Parenteral Nutrition Safety Committee Parenteral nutrition compatibility and stability: A comprehensive review. J. Parenter. Enter. Nutr. 2021, 46, 273–299. [Google Scholar] [CrossRef]
- Otero-Millán, L.; Rivero, N.L.; Rodicio, A.B.; Beloso, N.G.; Soto, J.L.L.; Piñeiro-Corrales, G. Stability of lipid emulsion in total parenteral nutrition: An overview of literature. Clin. Nutr. ESPEN 2021, 45, 19–25. [Google Scholar] [CrossRef]
- Watrobska-Swietlikowska, D. Stability of commercial parenteral lipid emulsions repacking to polypropylene syringes. PLoS ONE 2019, 14, e0214451. [Google Scholar] [CrossRef]
- Driscoll, D.F. The pharmacopeial evolution of intralipid injectable emulsion in plastic containers: From a coarse to a fine dispersion. Int. J. Pharm. 2009, 368, 193–198. [Google Scholar] [CrossRef]
- Gonyon, T.; Tomaso, A.E.; Kotha, P.; Owen, H.; Patel, D.; Carter, P.W.; Cronin, J.; Green, J.B.D. Interactions between Parenteral Lipid Emulsions and Container Surface. PDA J. Pharm. Sci. Tech. 2013, 67, 247–254. [Google Scholar] [CrossRef]
- Driscoll, D.F.; Ling, P.-R.; Bistrian, B.R. Pharmacopeial compliance of fish oil-containing parenteral lipid emulsion mixtures: Globule size distribution (GSD) and fatty acid analyses. Int. J. Pharm. 2009, 379, 125–130. [Google Scholar] [CrossRef]
- Hippalgaonkar, S.; Majumdar, S.; Kansara, V. Injectable Lipid Emulsions—Advancements, Opportunities and Challenges. AAPS PharmaSciTech 2010, 11, 1526–1540. [Google Scholar] [CrossRef]
- Forchielli, M.; Bonoli, A.; Bruno, L.; Piro, F.; Stancari, A.; Guarguaglini, A.; Maselli, S.; Pession, A.; Puggioli, C.; Bersani, G. Physicochemical stability assessment of parenteral nutrition admixtures for pediatric patients compounded with different kinds of fish oil-based emulsion. Nutrition 2016, 32, 403. [Google Scholar] [CrossRef]
- Forchielli, M.; Bonoli, A.; Preite, I.; Stancari, A.; Maselli, S.; Guarguaglini, A.; Mignini, I.; Masi, M.; Puggioli, C.; Bersani, G. Parenteral nutrition admixtures for pediatric patients compounded with highly refined fish oil-based emulsion: Assessment of physicochemical stability. Clin. Nutr. 2014, 33, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Télessy, I.G.; Balogh, J.; Szabó, B.; Csempesz, F.; Zelkó, R. Kinetic stability of all-in-one parenteral nutrition admixtures in the presence of high dose Ca2+ additive under clinical application circumstances. Nutr. J. 2012, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Pietka, M.; Klek, S. Parenteral nutrition admixtures for pediatric patients compounded with highly refined fish oil-based emulsion: Assessment of physicochemical stability—Letter to Editor. Clin. Nutr. 2015, 34, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Pietka, M.; Watrobska-Swietlikowska, D.; Szczepanek, K.; Szybinski, P.; Sznitowska, M. Nutritional support teams: The cooperation among physicians and pharmacists helps improve cost-effectiveness of home parenteral nutrition (HPN). NutrHosp 2015, 31, 251–259. [Google Scholar]
- Sandle, T. Manufacturing and validation, Pharmaceutical Microbiology, 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 2015; Volume 1, pp. 247–255. ISBN 978-0-08-100044-1. [Google Scholar]
- Kanuri, P. Science and risk based approach to the process validation—Link from quality by design to process validation. Int. J. Pharm. Sci. Res. 2016, 7, 914–929. [Google Scholar]
- Abdellah, A.; Noordin, M.I.; Ismail, W.A.W. Importance and globalization status of good manufacturing practice (GMP) requirements for pharmaceutical excipients. Saudi Pharm. J. 2013, 23, 9–13. [Google Scholar] [CrossRef]
- Sandle, T.; Lambda, S.S. Effectively incorporating quality risk management into quality systems. In Achieving Quality and Compliance Excellence in Pharmaceuticals: A Master Class GMP Guide; Saghee, M., Ed.; Business Horizons: New Delhi, India, 2012; pp. 89–128. [Google Scholar]
- Muselík, J.; Franc, A.; Doležel, P.; Goněc, R.; Krondlová, A.; Lukášová, I. Influence of Process Parameters on Content Uniformity of a Low Dose Active Pharmaceutical Ingredient in a Tablet Formulation According to GMP. Acta Pharm. 2014, 64, 355–367. [Google Scholar] [CrossRef]
- BRiley, S.; Li, X. Quality by Desing and Process Analytical Technology for Sterile Products—Where are we now? AAPS Pharm. Sci. Tech. 2011, 12, 114–118. [Google Scholar] [CrossRef]
- Chaudhari, R.A.; Ashok, P.P.; Chetan, S.C.; Chetan, Y.; Kantilal, A.P. Quality by design (QbD): An emerging paradigm for development of Pharmaceuticals. Int. J. Pharm. 2014, 4, 138–146. [Google Scholar]
- The World Health Organization (WHO). The International Pharmacopeia, 10th ed.; Parenteral Preparations. 2020. Available online: https://digicollections.net/phint/2020/index.html#d/b (accessed on 3 May 2022).
- Brooker, C. Mosby’s Dictionary of Medicine. Edinburgh: Nursing and Health Professions; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Sandle, T. Regulatory requirements and Good Manufacturing Practices (GMP). In Sterility, Sterilisation and Sterility Assurance for Pharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2013; pp. 35–54. [Google Scholar] [CrossRef]
- Outterson, K. Perspective: Regulation compounding pharmacies after NECC. New Engl. J. Med. 2012, 367, 1969–1972. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Huisman, I.; Roos, P.; Rietveld, A.; Klein, J.; Harbers, J.; Dorresteijn, J.; van Steenbergen, J.; Vos, M. Outbreak of severe sepsis due to contaminated propofol: Lessons to learn. J. Hosp. Infect. 2010, 76, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; Sheikhbabayi, M.; Samadi, N.; Jamalifar, H.; Dalili, D.; Samadi, N. Bacterial contamination of single- and multiple-dose vials after multiple use and intravenous admixtures in three different hospitals in Iran. Iran. J. Pharm. Res. 2013, 12, 205–209. [Google Scholar] [PubMed]
- Zorrilla-Veca, A.; Escandón-Vargas, K.; Brand-Giraldo, V.; León, T.; Herrera, M.; Payán, A. Bacterial contamination of propofol vials used in operating rooms of a third-level hospital. Am. J. Infect. Control. 2016, 44, e1–e3. [Google Scholar] [CrossRef]
- De la, T.; Huertas-Jimenez, A.; Silva, Y.; Gonzales, L.; Polanco, C. Endemic IV fluid contamination in hospitalized children in Mexico. A problem of serious public health consequences. Am. J. Infect. Control. 2012, 40, e177–e199. [Google Scholar]
- Centers of Disease Control and Prevention. Notes from the field: Multistate outbreak of postprocedural fungal endophthalmitis associated with a single compounding pharmacy—United States. Morb. Mortal. Wkly 2012, 61, 310–311. [Google Scholar]
- Ping-Cherng, C.; Wu, T.L.; Kuo, A.J.; Huang, Y.C.; Chung, T.Y.; Lin, C.S.; Leu, H.S.; Su, L.H. Outbreak of Serratia marcescens postsurgical bloodstream infection due to contaminated intravenous pain control fluids. Int. J. Infect. Dis. 2013, 17, e718–e722. [Google Scholar]
- Boseley, S. Questions Remain over Source of ITH Pharma Baby Feed Contamination. The Guardian. Available online: http://www.theguardian.com/society/2014/jun/05/questions-remain-source-ith-pharma-baby-feed-contamination (accessed on 5 June 2014).
- Austin, P.; Hand, K.; Elia, M. Systematic review and meta-analysis of the risk of microbial contamination of parenteral doses prepared under aseptic techniques in clinical and pharmaceutical environments: An update. J. Hosp. Infect. 2015, 91, 306–318. [Google Scholar] [CrossRef]
- Larmené-Beld, K.H.M.; Frijlink, H.W.; Taxis, K. A systematic review and meta-analysis of microbial contamination of parenteral medication prepared in a clinical versus pharmacy environment. Eur. J. Clin. Pharmacol. 2019, 75, 609–617. [Google Scholar] [CrossRef]
- Sandle, T. Cleanrooms, Isolators and Cleanroom Technology, Sterility, Sterilisation and Sterility Assurance for Pharmaceuticals; Woodhead Publishing: Cambridge, UK, 2013; pp. 189–207. [Google Scholar] [CrossRef]
- European Commission. Eudralex. The Rules Governing Medicinal Products in the European Community, Annex 1; European Commission: Brussels, Belgium, 2009. [Google Scholar]
- Sandle, T. Sterility, Sterilisation and Sterility Assurance for Pharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Gopinath, E.; Bhadauria, R.S.; Mishra, A.; Soan, V.K.; Gupta, D.P. Aseptic processing risk management: A review. IJPSR 2010, 1, 30–40. [Google Scholar]
- Haag, M. Calculating and Understanding Particulate Contamination Risk. Pharm. Tech. Eur. 2011, 23, 38–41. [Google Scholar]
- Vernjans, B.; Reed, C.H. Assessing filling technologies for contamination risk. BioPharm Int. 2012, 25, 3. [Google Scholar]
- Mahdiani, H.; Doroud, D.; Shahali, M.; Pouyan, N.; Brufar, F.; Rahimi, A.; Vaez, J. A practical risk-based approach to assess vial’s dimensions deviations effect on the aseptic filling processing, according to ICH Q9 guideline. Int. J. Pharm. Pharm. Sci. 2014, 6, 613–616. [Google Scholar]
- The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), ICH HARMONISED GUIDELINE. ICH Guideline Q9 (R1) on Quality Risk Management. 2021. Available online: https://www.gmp-navigator.com/files/guidemgr/ICHQ9.pdf (accessed on 4 May 2022).
- Domokos, A.; Pusztai, É.; Madarász, L.; Nagy, B.; Gyürkés, M.; Farkas, A.; Fülöp, G.; Casian, T.; Szilágyi, B.; Nagy, Z.K. Combination of PAT and mechanistic modeling tools in a fully continuous powder to granule line: Rapid and deep process understanding. Powder Technol. 2021, 388, 70–81. [Google Scholar] [CrossRef]
- Shah, E.B.; Gajjar, A. Process Analytical Technology (PAT) in Quality Assurance: A Detailed Review. Int. J. Pharm. Sci. Nanotechnol. 2022, 15, 5763–5770. [Google Scholar] [CrossRef]
- Mali, A.; Jagtap, M.; Karekar, P.; Maruska, A. A Brief review on process analytical (PAT) Review Article. Int. J. Curr. Pharm. Rev. Res. 2016, 8, 10–15. [Google Scholar]
- Regier, M.; Schubert, H.; Schuchmann, H. Dielectric spectroscopy—A new method for particle size- and fraction-determination. Innov. Food Sci. Emerg. Technol. 2004, 5, 199–204. [Google Scholar] [CrossRef]
- Regier, M. Überdielektrische und Magnetresonanz-MethodenzurCharakterisierung Disperser Systeme: Dissertation; Universität Karlsruhe: Berlin, Germany, 2003. [Google Scholar]
- Asami, K. Characterization of heterogeneous systems by dielectric spectroscopy. Prog. Polym. Sci. 2002, 27, 1617–1659. [Google Scholar] [CrossRef]
- Stuckly, S.S.; Gregory, G.; Anderson, L.; Kraszewski, A. A new sensor for dielectric measurements. IEEE Trans. Instrum. Meas. 1986, 35, 138–141. [Google Scholar] [CrossRef]
- Regier, M.; Schuchmann, H.P.; Danner, T. Online-Partikelgrößenmessung mittels dielektrischer Spektroskopie: Einsatzmöglichkeiten und Grenzen. Chem. Ing. Tech. 2006, 6, 78. [Google Scholar] [CrossRef]
- Beer, S.; Dobler, D.; Gross, A.; Ost, M.; Elseberg, C.; Maeder, U.; Schmidts, T.M.; Keusgen, M.; Fiebich, M.; Runkel, F. In line monitoring of the preparation of water-in-oil-in-water (W/O/W) type multiple emulsions via dielectric spectroscopy. Int. J. Pharm. 2013, 441, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Volker, A.; de Kroon, M. Characterization of suspensions by ultrasonic reflection measurements. Ultrasonics 1998, 36, 283–289. [Google Scholar] [CrossRef]
- Weser, R.; Wöckel, S.; Wessely, B.; Hempel, U. Particle characterisation in highly concentrated dispersions using ultrasonic backscattering method. Ultrasonics 2012, 53, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Faran, J.J. Sound Scattering by Solid Cylinders and Spheres. J. Acoust. Soc. Am. 1951, 23, 405–418. [Google Scholar] [CrossRef]
- Weser, R.; Wöckel, S.; Hempel, U.; Wessely, B.; Auge, J. Particle characterization in highly concentrated suspensions by ultrasound scattering method. Sens. Actuators A 2013, 202, 30–36. [Google Scholar] [CrossRef]
- Weser, R.; Woeckel, S.; Wessely, B.; Steinmann, U.; Babick, F.; Stintz, M. Ultrasonic backscattering method for in-situ characterisation of concentrated dispersions. Powder Technol. 2014, 268, 177–190. [Google Scholar] [CrossRef]
- Dukhin, A.S.; Goetz, P.J. Acoustic and electroacoustic spectroscopy for characterizing concentrated dispersions and emulsions. Adv. Colloid Interface Sci. 2001, 92, 73–132. [Google Scholar] [CrossRef]
- Dukhin, A.S.; Goetz, P.J. Ultrasound for Characterizing Colloids; Elsevier: Amsterdam, The Netherlands, 2004; pp. 91–119. [Google Scholar] [CrossRef]
- Wilhelm, P.; Stephan, D. On-line tracking of the coating of nanoscaled silica with titania nanoparticles via zeta-potential measurements. Colloid Interface Sci. 2006, 293, 88–92. [Google Scholar] [CrossRef]
- Dukhin, A.S.; Goetz, P.J.; Xiaohua, F.; Somasundaran, P. Monitoring nanoparticles in the presence of larger particles in liquids using acoustics and electron microscopy. J. Colloid Interface Sci. 2010, 342, 18–25. [Google Scholar] [CrossRef]
- Purohit, P.S.; Kulkarni, R.; Somasundaran, P. Investigation of colloidal properties of modified silicone polymers emulsified by non-ionic surfactants. J. Colloid Interface Sci. 2012, 383, 49–54. [Google Scholar] [CrossRef]
- DosRamos, J.G. Acoustic attenuation spectroscopy for process control of dispersed systems. IOP Conf. Series Mater. Sci. Eng. 2012, 42. [Google Scholar] [CrossRef]
- Ding, B.; He, L.P.; Luo, J.H.; Peng, B.L.; Geng, X.F.; Wang, P.M. Determination of particle size of heavy oil in water dispersion system by ultra sonic attenuation method. J. Pet. Sci. Eng. 2016, 146, 764–769. [Google Scholar] [CrossRef]
- Cao, Y.; Jin, Y.; Li, J.; Zou, D.; Chen, X. Demulsification of the phosphoric acid–tributyl phosphate (W/O) emulsion by hydrocyclone. Sep. Purif. Technol. 2016, 158, 387–395. [Google Scholar] [CrossRef]
- Silva, C.A.; Saraiva, S.V.; Bonetti, D.; Higuti, R.T.; Cunha, R.L.; Pereira, L.O.; Silva, F.V.; Fileti, A.M. Measurements of bimodal droplet size distribution of emulsions using ultrasonic spectroscopy in the long and intermediate wavelength regimes. Chem. Eng. Sci. 2021, 252, 117274. [Google Scholar] [CrossRef]
- Weiwei, E.; Jingcai, C.; Chao, Y.; Zaisha, M. Experimental study by online measurement of the precipitation of nickel hydroxide: Effect of operating conditions. Chin. J. Chem. Eng. 2015, 23, 860–867. [Google Scholar]
- Pinto, J.T.; Faulhammer, E.; Dieplinger, J.; Dekner, M.; Makert, C.; Nieder, M.; Paudel, A. Progress in spray-drying of protein pharmaceuticals: Literature analysis of trends in formulation and process attributes. Dry. Technol. 2021, 39, 1415–1446. [Google Scholar] [CrossRef]
- Medendorp, J.; Bric, J.; Connelly, G.; Tolton, K.; Warman, M. Development and beyond: Strategy for long-term maintenance of an online laser diffraction particle size method in a spray drying manufacturing process. J. Pharm. Biomed. Anal. 2015, 112, 79–84. [Google Scholar] [CrossRef]
- Sabin, A. Problems in particle size: Laser diffraction observations. GXP J. 2011, 15, 35–44. [Google Scholar]
- Reufer, M.; Machado, A.H.; Niederquell, A.; Bohnenblust, K.; Müller, B.; Völker, A.C.; Kuentz, M. Introducing Diffusing Wave Spectroscopy as a Process Analytical Tool for Pharmaceutical Emulsion Manufacturing. J. Pharm. Sci. 2014, 103, 3902–3913. [Google Scholar] [CrossRef]
- Kozdrach, R. The Innovative Research Methodology of Tribological and Rheological Properties of Lubricating Grease. Tribol. Ind. 2021, 43, 117–130. [Google Scholar] [CrossRef]
- Kolman, M.; Boland, G.; Amin, S. Exploring the Utility of Diffusing Wave Spectroscopy (DWS) as a Novel Tool for Early Detection of Stability Issues in Cosmetic Emulsions. Cosmetics 2021, 8, 99. [Google Scholar] [CrossRef]
- Chen, Z.; Peng, J.; Ge, L.; Xu, Z. Demulsifying water-in-oil emulsions by ethyl cellulose demulsifiers studied using focused beam reflectance measurement. Chem. Eng. Sci. 2015, 130, 254–263. [Google Scholar] [CrossRef]
- Dowding, P.J.; Goodwin, J.W.; Vincent, B. Factors governing emulsion droplet and solid particle size measurements performed using the focused beam reflectance technique. Colloids Surfaces A: Physicochem. Eng. Asp. 2001, 192, 5–13. [Google Scholar] [CrossRef]
- Li, H.; Grover, M.A.; Kawajiri, Y.; Rousseau, R.W. Development of an empirical method relating crystal size distributions and FBRM measurements. Chem. Eng. Sci. 2013, 89, 142–151. [Google Scholar] [CrossRef]
- Barros, L.; Gim-Krumm, M.; Seriche, G.; Quilaqueo, M.; Castillo, C.; Ihle, C.F.; Ruby-Figueroa, R.; Estay, H. In-situ and real-time aggregation size evolution of copper sulfide precipitates using focused beam reflectance measurement (FBRM). Powder Technol. 2020, 380, 205–218. [Google Scholar] [CrossRef]
- Less, S.; Vilagines, R. Light beam reflectance measurement of droplets diameter distribution in crude oil emulsions. Fuel 2013, 109, 542–550. [Google Scholar] [CrossRef]
- Less, S.; Vilagines, R.; Al-Khalifa, H. Online droplet diameter measurements to improve the crude oil dehydration process. Chem. Eng. Res. Des. 2014, 92, 2659–2667. [Google Scholar] [CrossRef]
- Cobbledick, J.; Nguyen, A.; Latulippe, D.R. Demonstration of FBRM as process analytical technology tool for dewatering processes via CST correlation. Water Res. 2014, 58, 132–140. [Google Scholar] [CrossRef]
- Ruf, A.; Worlitschek, J.; Mazzotti, M. Modeling and Experimental Analysis of PSD Measurements through FBRM. Part. Part. Syst. Charact. 2000, 17, 167–179. [Google Scholar] [CrossRef]
- Greaves, D.; Boxall, J.; Mulligan, J.; Montesi, A.; Creek, J.; Sloan, E.D.; Koh, C.A. Measuring the particle size of a known distribution using the focused beam reflactancemeasurment technique. Chem. Eng. Sci. 2008, 63, 5410–5419. [Google Scholar] [CrossRef]
- Heath, A.R.; Fawell, P.D.; Bahri, P.A.; Swift, J.D. Estimating Average Particle Size by Focused Beam Reflectance Measurement (FBRM). Part. Part. Syst. Charact. 2002, 19, 84–95. [Google Scholar] [CrossRef]
- Meng, X.; Qi, L.; Zhang, R.; Liu, H.; Xu, C.; Liu, Z.; Klusener, P.A. Droplet size distribution and droplet size correlation of chloroaluminate ionic liquid-heptane dispersion in a stirred vessel. Chem. Eng. J. 2015, 268, 116–124. [Google Scholar]
- Liu, J.; Wang, W.; Wang, P.Y.; Duan, J.M.; Gong, J. Evolution of dispersed drops during the mixing of mineral oil and water phases in a stirring tank. Chem. Eng. Sci. 2013, 91, 173–179. [Google Scholar]
- Calabrese, R.V.; Rueger, P.E. Dispersion of water into oil in a rotor-stator mixer. Part 1: Drop breakup in dilute systems. Chem. Eng. Des. 2013, 91, 2122–2133. [Google Scholar]
- Melchuna, A.; Cameirao, A.; Herri, J.-M.; Glenat, P. Topological modeling of methane hydrate crystallization from low to high water cut emulsion systems. Fluid Phase Equilibria 2016, 413, 158–169. [Google Scholar] [CrossRef]
- Turner, D.; Miller, K.; Sloan, E. Direct conversion of water droplets to methane hydrate in crude oil. Chem. Eng. Sci. 2009, 64, 5066–5072. [Google Scholar] [CrossRef]
- Mirshekari, F.; Pakzad, L.; Fatehi, P. An investigation on the stability of the hazelnut oil-water emulsion. J. Dispers. Sci. Technol. 2019, 41, 929–940. [Google Scholar] [CrossRef]
- Desnoyer, C.; Masbernat, O.; Gourdon, C. Experimental study of drop size distributions at high phase ratio in liquid–liquid dispersions. Chem. Eng. Sci. 2003, 58, 1353–1363. [Google Scholar] [CrossRef]
- El-Hamouz, A.; Cooke, M.; Kowalski, A.; Sharratt, P. Dispersion of Silicone Oil in Water Surfactant Solution: Effect on Impeller Speed, Oil Viscosity and Addition Point of Drop Size Distribution. Chem. Eng. Processing 2009, 48, 633–642. [Google Scholar] [CrossRef]
- Muhaimin, M.; Chaerunisaa, A.Y.; Bodmeier, R. Real-time particle size analysis using focused beam reflectance measurement as a process analytical technology tool for continuous microencapsulation process. Sci. Rep. 2021, 11, 19390. [Google Scholar] [CrossRef]
- Acevedo, D.; Wu, W.-L.; Yang, X.; Pavurala, N.; Mohammad, A.; O’Connor, T.F. Evaluation of focused beam reflectance measurement (FBRM) for monitoring and predicting the crystal size of carbamazepine in crystallization processes. CrystEngComm 2020, 23, 972–985. [Google Scholar] [CrossRef]
- Maaß, S.; Wollny, S.; Voigt, A.; Kraume, M. Experimental comparison of measurement techniques for drop size distributions in liquid/liquid dispersions. Exp. Fluids 2010, 50, 259–269. [Google Scholar] [CrossRef]
- Maaß, S.; Rojahn, J.; Hänsch, R.; Kraume, M. Automated drop detection using image analysis for online particle size monitoring in multiphase systems. Comput. Chem. Eng. 2012, 45, 27–37. [Google Scholar] [CrossRef]
- Maaß, S.; Paul, N.; Kraume, M. Influence of the dispersed phase fraction on experimental and predicted drop size distributions in breakage dominated stirred liquid-liquid systems. Chem. Eng. Sci. 2012, 76, 140–153. [Google Scholar] [CrossRef]
- Maaß, S.; Rehm, T.; Kraume, M. Prediction of drop sizes for liquid–liquid systems in stirred slim reactors—Part II: Multi stage impellers. Chem. Eng. J. 2011, 168, 827–838. [Google Scholar] [CrossRef]
- Khakpay, A.; Abolghasemi, H. The effects of impeller speed and holdup on mean drop size in a mixer settler with spiral-type impeller. Can. J. Chem. Eng. 2010, 88, 329–334. [Google Scholar] [CrossRef]
- Razzaghi, K.; Shahraki, F. On the effect of phase fraction on drop size distribution of liquid–liquid dispersions in agitated vessels. Chem. Eng. Res. Des. 2010, 88, 803–808. [Google Scholar] [CrossRef]
- Vancleef, A.; Seurs, S.; Jordens, J.; Van Gerven, T.; Thomassen, L.C.J.; Braeken, L. Reducing the Induction Time Using Ultrasound and High-Shear Mixing in a Continuous Crystallization Process. Crystals 2018, 8, 326. [Google Scholar] [CrossRef]
- Domokos, A.; Madarász, L.; Stoffán, G.; Tacsi, K.; Galata, D.; Csorba, K.; Vass, P.; Nagy, Z.K.; Pataki, H. Real-Time Monitoring of Continuous Pharmaceutical Mixed Suspension Mixed Product Removal Crystallization Using Image Analysis. Org. Process. Res. Dev. 2021, 26, 149–158. [Google Scholar] [CrossRef]
- Dieter, P.; Stefan, D.; Günter, E.; Michael, K. In-line particle sizing for real-time process control by fibre-optical spatial filtering technique (SFT). Adv. Powder Technol. 2011, 22, 203–208. [Google Scholar] [CrossRef]
- Burggraeve, A.; Kerkhof, T.V.D.; Hellings, M.; Remon, J.; Vervaet, C.; De Beer, T. Evaluation of in-line spatial filter velocimetry as PAT monitoring tool for particle growth during fluid bed granulation. Eur. J. Pharm. Biopharm. 2010, 76, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Roßteuscher-Carl, K.; Fricke, S.; Hacker, M.C.; Schulz-Siegmund, M. In-line monitoring of particle size in a fluid bed granulator: Investigations concerning positioning and configuration of the sensor. Int. J. Pharm. 2014, 466, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Vancleef, A.; Maes, D.; Van Gerven, T.; Thomassen, L.C.; Braeken, L. Flow-through microscopy and image analysis for crystallization processes. Chem. Eng. Sci. 2021, 248, 117067. [Google Scholar] [CrossRef]
- Emmerich, J.; Tang, Q.; Wang, Y.; Neubauer, P.; Junne, S.; Maaß, S. Optical inline analysis and monitoring of particle size and shape distributions for multiple applications: Scientific and industrial relevance. Chin. J. Chem. Eng. 2018, 27, 257–277. [Google Scholar] [CrossRef]
- Peña, R.; Jarmer, D.J.; Burcham, C.; Nagy, Z.K. Further Understanding of Agglomeration Mechanisms in Spherical Crystallization Systems: Benzoic Acid Case Study. Cryst. Growth Des. 2019, 19, 1668–1679. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measurement Technique | Advantages | Disadvantages | Commercially Available |
|---|---|---|---|
| Dielectric spectroscopy | Measurement of concentrated emulsions possible; no dilution needed, fast measurement, | Measurement is influenced by temperature and conductivity fluctuations, bimodalities affect the spectra; product specific calibration needed | No for emulsions. Available for biological cells from Aber Instruments Ltd. and Hamilton Company |
| Ultrasonic backscattering | Measurement of concentrated emulsions; no dilution needed; | Material parameters should be well known; not suitable for nano particles | No online/inline system for emulsions available |
| Ultrasonic attenuation spectroscopy | Measurement of concentrated emulsions; no dilution needed; wide measuring range 1 µm–3 mm possible | Requires data for phase concertation; not suitable for nano particles | Online sensor available for emulsions from Dispersion Technology Inc. (DT-1202); Inline sensor available from Sympatec GmbH (OPUS) |
| Laser diffraction | No calibration needed wide measuring range 0.1 µm–1 mm possible; fast measurement; | Not suitable for nano particles | Available for emulsions from Malvern Panalytical Ltd. (Insitec L and SX) |
| Diffusing wave spectroscopy | No product specific calibration required; suitable for nano particles | No inline technique; dilution of the sample required | Available for emulsions from Microtrac Retsch GmbH (Nanotrac Flex) |
| Laser scanner technology | No product specific calibration required, suitable for nano particles; wide measurement range 0.5–2 µm | Requires sufficient material flows to provide valid data | Available for emulsions from Mettler Toledo GmbH (FBRM) and Sequip S + E GmbH (ECA) |
| Spatial filtering technique | No product specific calibration required; wide measurement range 50 µm–6 mm | Only suitable for low concentrations | No for emulsions. Available for powders from Parsum GmbH (IPP 70-S) |
| Insitu microscope | Single large droplets detectable; information about particle shape; wide measurement range 5 µm–10 mm | Generates large amounts of image data (data backup is a challenge) | Available for emulsions from SOPAT GmbH (SOPAT-VR) and Sequip S + E GmbH (IVM) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grumbach, C.; Czermak, P. Process Analytical Technology for the Production of Parenteral Lipid Emulsions According to Good Manufacturing Practices. Processes 2022, 10, 1174. https://doi.org/10.3390/pr10061174
Grumbach C, Czermak P. Process Analytical Technology for the Production of Parenteral Lipid Emulsions According to Good Manufacturing Practices. Processes. 2022; 10(6):1174. https://doi.org/10.3390/pr10061174
Chicago/Turabian StyleGrumbach, Carsten, and Peter Czermak. 2022. "Process Analytical Technology for the Production of Parenteral Lipid Emulsions According to Good Manufacturing Practices" Processes 10, no. 6: 1174. https://doi.org/10.3390/pr10061174
APA StyleGrumbach, C., & Czermak, P. (2022). Process Analytical Technology for the Production of Parenteral Lipid Emulsions According to Good Manufacturing Practices. Processes, 10(6), 1174. https://doi.org/10.3390/pr10061174