
Alterations of Epigenetic Regulators in Pancreatic Cancer and Their Clinical Implications



Abstract
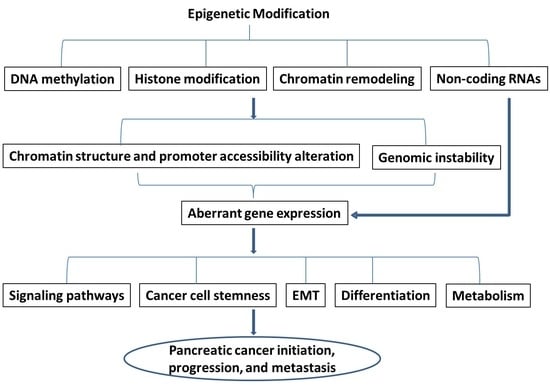
:
1. Introduction
2. DNA Methylation
3. Histone Modifications
3.1. Histone Methyltransferases and Demethylases
3.2. Histone Acetyltransferase and Deacetylase
3.3. Histone Ubiquitination
4. Chromatin Remodeling
5. Non-Coding RNA
5.1. Small RNAs
5.2. LncRNA
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| RNA | Ribonucleic acid |
| PDA | Pancreatic ductal adenocarcinoma |
| DNA | Deoxyribonucleic acid |
| DNMT | DNA methyltransferase |
| DNA | Deoxyribonucleic acid |
| SAM | S-adenosylmethionine |
| ncRNA | Non-coding RNA |
| piRNA | Piwi-interacting RNA |
| pRNA | Promoter-associated RNA |
| PanIN | Pancreatic intraepithelial neoplasia |
| IPMN | Intraductal papillary mucinous neoplasm |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| HMT | Histone methyltransferase |
| HDM | Histone demethylase |
| HKMT | Histone lysine methyltransferase |
| KDM | Lysine-specific demethylase |
| CBP | cAMP-response element binding protein |
| PRC1 | Polycomb repressive complex 1 |
| SWI/SNF | SWItch/sucrose non-fermentable |
| lncRNA | Long non-coding RNA |
| RNAi | RNA interference |
| MPC | Multipotent pancreatic progenitor cell |
| miRNA | microRNA |
References
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tsutsumi, S.; Kawaguchi, T.; Nagasaki, K.; Tatsuno, K.; Yamamoto, S.; Sang, F.; Sonoda, K.; Sugawara, M.; Saiura, A.; et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 2012, 22, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744–6755. [Google Scholar] [CrossRef] [PubMed]
- Quilichini, E.; Haumaitre, C. Implication of epigenetics in pancreas development and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.M.; Walsh, L.A.; Chan, T.A. Driver mutations of cancer epigenomes. Protein Cell 2014, 5, 265–296. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Denis, H.; Ndlovu, M.N.; Fuks, F. Regulation of mammalian DNA methyltransferases: A route to new mechanisms. EMBO Rep. 2011, 12, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Gowher, H.; Jeltsch, A. Enzymatic properties of recombinant DNMT3A DNA methyltransferase from mouse: The enzyme modifies DNA in a non-processive manner and also methylates of non-CpG sites. J. Mol. Boil. 2001, 309, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Li, Y.; Duan, S.; Bronner, C.; Arrowsmith, C.H.; Dhe-Paganon, S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 2008, 455, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. ChemBioChem Eur. J. Chem. Boil. 2011, 12, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of DNMT3A bound to DNMT3L suggests a model for de novo DNA methylation. Nature 2007, 449, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Smith, A.D.; Kendall, J.; Xuan, Z.; Ravi, K.; Rooks, M.; Zhang, M.Q.; Ye, K.; Bhattacharjee, A.; Brizuela, L.; et al. High definition profiling of mammalian DNA methylation by array capture and single molecule bisulfite sequencing. Genome Res. 2009, 19, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.A.; Sachidanandam, R.; Bourc’his, D.; Schaefer, C.; Pezic, D.; Toth, K.F.; Bestor, T.; Hannon, G.J. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 2008, 31, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, L.; Nakielny, S. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Boil. Chem. 2004, 279, 49479–49487. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M.S.; Villeneuve, L.M.; Ehsani, A.; Amarzguioui, M.; Aagaard, L.; Chen, Z.X.; Riggs, A.D.; Rossi, J.J.; Morris, K.V. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA 2006, 12, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3B and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Lu, P.J.; Ding, L.Y.; Chu, P.C.; Hsu, W.Y.; Chen, C.S.; Tsao, C.C.; Chen, B.H.; Lee, C.T.; Shan, Y.S.; et al. TGF-β promotes mesenchymal phenotype of pancreatic cancer cells, in part, through epigenetic activation of VAV1. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Kottakis, F.; Nicolay, B.N.; Roumane, A.; Karnik, R.; Gu, H.; Nagle, J.M.; Boukhali, M.; Hayward, M.C.; Li, Y.Y.; Chen, T.; et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 2016, 539, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Jia, Y.; Yu, Z.; House, M.G.; Esteller, M.; Brock, M.V.; Herman, J.G. Epigenetic changes associated with neoplasms of the exocrine and endocrine pancreas. Discov. Med. 2014, 17, 67–73. [Google Scholar] [PubMed]
- Nones, K.; Waddell, N.; Song, S.; Patch, A.M.; Miller, D.; Johns, A.; Wu, J.; Kassahn, K.S.; Wood, D.; Bailey, P.; et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and met signaling. Int. J. Cancer 2014, 135, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Guzzetta, A.A.; Bailey, V.J.; Downing, S.R.; Van Neste, L.; Chiappinelli, K.B.; Keeley, B.P.; Stark, A.; Herrera, A.; Wolfgang, C.; et al. Novel methylation biomarker panel for the early detection of pancreatic cancer. Clin. Cancer Res. 2013, 19, 6544–6555. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, S.D.; Madsen, P.H.; Larsen, A.C.; Johansen, M.B.; Drewes, A.M.; Pedersen, I.S.; Krarup, H.; Thorlacius-Ussing, O. Cell-free DNA promoter hypermethylation in plasma as a diagnostic marker for pancreatic adenocarcinoma. Clin. Epigenet. 2016, 8, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.S.; Bamlet, W.R.; Oberg, A.L.; de Andrade, M.; Matsumoto, M.E.; Tang, H.; Thibodeau, S.N.; Petersen, G.M.; Wang, L. Leukocyte DNA methylation signature differentiates pancreatic cancer patients from healthy controls. PLoS ONE 2011, 6, e18223. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gao, J.; Man, X.H.; Li, Z.S.; Gong, Y.F. Significance of DNA methyltransferase-1 and histone deacetylase-1 in pancreatic cancer. Oncol. Rep. 2009, 21, 1439–1447. [Google Scholar] [PubMed]
- Gao, J.; Wang, L.; Xu, J.; Zheng, J.; Man, X.; Wu, H.; Jin, J.; Wang, K.; Xiao, H.; Li, S.; et al. Aberrant DNA methyltransferase expression in pancreatic ductal adenocarcinoma development and progression. J. Exp. Clin. Cancer Res. 2013, 32, 86. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Wang, Y.F.; Li, G.; Yang, S.S.; Liu, C.; Hu, H.; Jin, G.; Hu, X.G. Interplay between MENIN and DNMT1 reversibly regulates pancreatic cancer cell growth downstream of the hedgehog signaling pathway. Cancer Lett. 2016, 370, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Zhu, Y.; Zhu, Y.; Wu, J.L.; Liang, W.B.; Zhu, R.; Xu, Z.K.; Du, Q.; Miao, Y. Association of increased DNA methyltransferase expression with carcinogenesis and poor prognosis in pancreatic ductal adenocarcinoma. Clin. Transl. Oncol. 2012, 14, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Mund, C.; Brueckner, B.; Lyko, F. Reactivation of epigenetically silenced genes by DNA methyltransferase inhibitors: Basic concepts and clinical applications. Epigenetics 2006, 1, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Ball, B.; Zeidan, A.; Gore, S.D.; Prebet, T. Hypomethylating agent combination strategies in myelodysplastic syndromes: Hopes and shortcomings. Leuk. Lymphoma 2016, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Hu, G.; Luo, C.; Liang, Z. DNA methyltransferase inhibitors: An updated patent review (2012–2015). Expert Opin. Ther. Pat. 2016, 26, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Susanto, J.M.; Colvin, E.K.; Pinese, M.; Chang, D.K.; Pajic, M.; Mawson, A.; Caldon, C.E.; Musgrove, E.A.; Henshall, S.M.; Sutherland, R.L.; et al. The epigenetic agents suberoylanilide hydroxamic acid and 5AZA2′ deoxycytidine decrease cell proliferation, induce cell death and delay the growth of MIAPACA2 pancreatic cancer cells in vivo. Int. J. Oncol. 2015, 46, 2223–2230. [Google Scholar] [PubMed]
- Wang, X.; Wang, H.; Jiang, N.; Lu, W.; Zhang, X.F.; Fang, J.Y. Effect of inhibition of MEK pathway on 5-AZA-deoxycytidine-suppressed pancreatic cancer cell proliferation. Genet. Mol. Res. 2013, 12, 5560–5573. [Google Scholar] [CrossRef] [PubMed]
- Wongtrakoongate, P. Epigenetic therapy of cancer stem and progenitor cells by targeting DNA methylation machineries. World J. Stem Cells 2015, 7, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the cytotoxic effects of PARP inhibitors with DNA demethylating agents—A potential therapy for cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Derissen, E.J.; Beijnen, J.H.; Schellens, J.H. Concise drug review: Azacitidine and decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Festuccia, C.; Marampon, F.; Popov, V.M.; Pestell, R.G.; Zani, B.M.; Tombolini, V. Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Mol. Cancer 2010, 9, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhang, C.Y. Histone modifying enzymes: Novel disease biomarkers and assay development. Expert Rev. Mol. Diagn. 2016, 16, 297–306. [Google Scholar] [CrossRef] [PubMed]
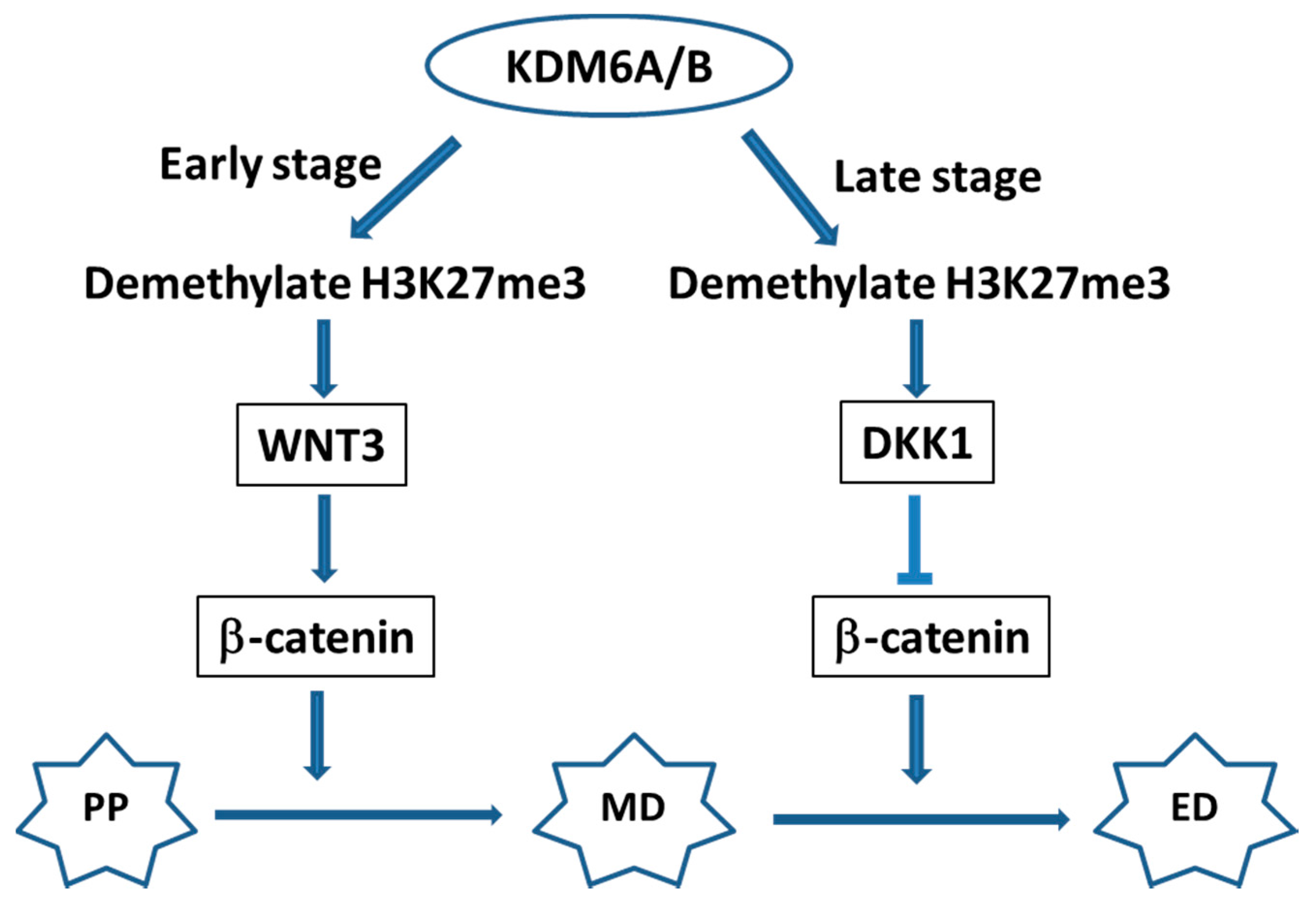
- Jiang, W.; Wang, J.; Zhang, Y. Histone H3K27ME3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCS by regulating WNT signaling pathway. Cell Res. 2013, 23, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Starmer, J.; Yee, D.; Pohlers, M.; Magnuson, T. KDM6 demethylase independent loss of histone H3 lysine 27 trimethylation during early embryonic development. PLoS Genet. 2014, 10, e1004507. [Google Scholar] [CrossRef] [PubMed]
- Von Figura, G.; Fukuda, A.; Roy, N.; Liku, M.E.; Morris Iv, J.P.; Kim, G.E.; Russ, H.A.; Firpo, M.A.; Mulvihill, S.J.; Dawson, D.W.; et al. The chromatin regulator BRG1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2014, 16, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of ZESTE homologue 2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef] [PubMed]
- Toll, A.D.; Dasgupta, A.; Potoczek, M.; Yeo, C.J.; Kleer, C.G.; Brody, J.R.; Witkiewicz, A.K. Implications of enhancer of ZESTE homologue 2 expression in pancreatic ductal adenocarcinoma. Hum. Pathol. 2010, 41, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Tang, A.J.; Castoreno, A.B.; Kuo, S.Y.; Wang, Q.; Kuballa, P.; Xavier, R.; Shamji, A.F.; Schreiber, S.L.; Wagner, B.K. Gossypol and an HMT G9a inhibitor act in synergy to induce cell death in pancreatic cancer cells. Cell Death Dis. 2013, 4, e690. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, T.; Wang, X.; Zhao, E.; Choi, J.H.; Yang, L.; Zha, Y.; Dong, Z.; Huang, S.; Asara, J.M.; et al. The histone H3 methyltransferase G9a epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013, 18, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.T.; So, C.W. Epigenetic therapies by targeting aberrant histone methylome in AML: Molecular mechanisms, current preclinical and clinical development. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Crea, F.; Paolicchi, E.; Funel, N.; Galvani, E.; Marquez, V.E.; Honeywell, R.J.; Danesi, R.; Peters, G.J.; Giovannetti, E. Molecular mechanisms involved in the synergistic interaction of the EZH2 inhibitor 3-deazaneplanocin a with gemcitabine in pancreatic cancer cells. Mol. Cancer Ther. 2012, 11, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Trojer, P. Targeting histone lysine methylation in cancer. Pharmacol. Ther. 2015, 150, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Kruidenier, L.; Chung, C.W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective JUMONJI H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, B.; Nielsen, J.M.; Hudlebusch, H.R.; Lees, M.J.; Larsen, D.V.; Boesen, T.; Labelle, M.; Gerlach, L.O.; Birk, P.; Helin, K. Inhibition of demethylases by Gsk-J1/J4. Nature 2014, 514, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Thinnes, C.C.; England, K.S.; Kawamura, A.; Chowdhury, R.; Schofield, C.J.; Hopkinson, R.J. Targeting histone lysine demethylases—Progress, challenges, and the future. Biochim. Biophys. Acta 2014, 1839, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Q.; Paulk, J.; Kubicek, S.; Kemp, M.M.; Adams, D.J.; Shamji, A.F.; Wagner, B.K.; Schreiber, S.L. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Boil. 2012, 7, 1152–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Mees, S.T.; Mardin, W.A.; Wendel, C.; Baeumer, N.; Willscher, E.; Senninger, N.; Schleicher, C.; Colombo-Benkmann, M.; Haier, J. EP300—A miRNA-regulated metastasis suppressor gene in ductal adenocarcinomas of the pancreas. Int. J. Cancer 2010, 126, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, P.; Seidler, B.; Schuler, S.; Schnieke, A.; Gottlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Ouaissi, M.; Sielezneff, I.; Silvestre, R.; Sastre, B.; Bernard, J.P.; Lafontaine, J.S.; Payan, M.J.; Dahan, L.; Pirro, N.; Seitz, J.F.; et al. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann. Surg. Oncol. 2008, 15, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Liang, I.C.; Yee, N.S. Histone deacetylase 1 is required for exocrine pancreatic epithelial proliferation in development and cancer. Cancer Boil. Ther. 2011, 11, 659–670. [Google Scholar] [CrossRef]
- Schneider, G.; Kramer, O.H.; Schmid, R.M.; Saur, D. Acetylation as a transcriptional control mechanism-HDACs and hats in pancreatic ductal adenocarcinoma. J. Gastrointest. Cancer 2011, 42, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.; Denkert, C.; Budczies, J.; Buckendahl, A.C.; Darb-Esfahani, S.; Noske, A.; Muller, B.M.; Bahra, M.; Neuhaus, P.; Dietel, M.; et al. High class I HDAC activity and expression are associated with RELA/P65 activation in pancreatic cancer in vitro and in vivo. BMC Cancer 2009, 9, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, C.; Goder, A.; Beyer, M.; Kiweler, N.; Mahendrarajah, N.; Rauch, A.; Nikolova, T.; Stojanovic, N.; Wieczorek, M.; Reich, T.R.; et al. Class I HISTONE deacetylases regulate P53/NF-κB crosstalk in cancer cells. Cell. Signal. 2017, 29, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.A.; Iovanna, J.; Urrutia, R. The promise of epigenomic therapeutics in pancreatic cancer. Epigenomics 2016, 8, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Neureiter, D.; Jager, T.; Ocker, M.; Kiesslich, T. Epigenetics and pancreatic cancer: Pathophysiology and novel treatment aspects. World J. Gastroenterol. 2014, 20, 7830–7848. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Farria, A.; Li, W.; Dent, S.Y. Kats in cancer: Functions and therapies. Oncogene 2015, 34, 4901–4913. [Google Scholar] [CrossRef] [PubMed]
- Marsoni, S.; Damia, G.; Camboni, G. A work in progress: The clinical development of histone deacetylase inhibitors. Epigenetics 2008, 3, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Bednar, F.; Schofield, H.K.; Collins, M.A.; Yan, W.; Zhang, Y.; Shyam, N.; Eberle, J.A.; Almada, L.L.; Olive, K.P.; Bardeesy, N.; et al. BMI1 is required for the initiation of pancreatic cancer through an INK4A-independent mechanism. Carcinogenesis 2015, 36, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Benitz, S.; Regel, I.; Reinhard, T.; Popp, A.; Schaffer, I.; Raulefs, S.; Kong, B.; Esposito, I.; Michalski, C.W.; Kleeff, J. Polycomb repressor complex 1 promotes gene silencing through H2AK119 mono-ubiquitination in acinar-to-ductal metaplasia and pancreatic cancer cells. Oncotarget 2016, 7, 11424–11433. [Google Scholar] [PubMed]
- Chen, S.; Chen, J.; Zhan, Q.; Zhu, Y.; Chen, H.; Deng, X.; Hou, Z.; Shen, B.; Chen, Y.; Peng, C. H2AK119UB1 and H3K27ME3 in molecular staging for survival prediction of patients with pancreatic ductal adenocarcinoma. Oncotarget 2014, 5, 10421–10433. [Google Scholar] [CrossRef] [PubMed]
- Meas, R.; Mao, P. Histone ubiquitylation and its roles in transcription and DNA damage response. DNA Repair 2015, 36, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Weake, V.M.; Workman, J.L. Histone ubiquitination: Triggering gene activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Goldknopf, I.L.; Busch, H. Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate-protein A24. Proc. Natl. Acad. Sci. USA 1977, 74, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Nickel, B.E.; Davie, J.R. Structure of polyubiquitinated histone H2A. Biochemistry 1989, 28, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, M. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 1996, 30, 405–439. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006, 7, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, G. A new way forward in cancer drug discovery: Inhibiting the SWI/SNF chromatin remodelling complex. ChemBioChem Eur. J. Chem. Biol. 2016, 17, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Giacomini, C.P.; Matsukuma, K.; Karikari, C.A.; Bashyam, M.D.; Hidalgo, M.; Maitra, A.; Pollack, J.R. Convergent structural alterations define switch/sucrose nonfermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Segedi, M.; Anderson, L.N.; Espin-Garcia, O.; Borgida, A.; Bianco, T.; Cheng, D.; Chen, Z.; Patel, D.; Brown, M.C.; Xu, W.; et al. BRM polymorphisms, pancreatic cancer risk and survival. Int. J. Cancer 2016, 139, 2474–2481. [Google Scholar] [CrossRef] [PubMed]
- Numata, M.; Morinaga, S.; Watanabe, T.; Tamagawa, H.; Yamamoto, N.; Shiozawa, M.; Nakamura, Y.; Kameda, Y.; Okawa, S.; Rino, Y.; et al. The clinical significance of SWI/SNF complex in pancreatic cancer. Int. J. Oncol. 2013, 42, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Tian, J.; Zhong, R.; Tian, Y.; Chen, W.; Qian, J.; Zou, L.; Xiao, M.; Shen, N.; Yang, H.; et al. Genetic variants in the SWI/SNF complex and smoking collaborate to modify the risk of pancreatic cancer in a Chinese population. Mol. Carcinog. 2015, 54, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A. Molecular mechanism of intraductal papillary mucinous neoplasm and intraductal papillary mucinous neoplasm-derived pancreatic ductal adenocarcinoma. J. Hepatobiliary Pancreat. Sci. 2015, 22, 519–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, B.; Guo, M.; Reynolds, A.; Hara, M.; Stein, R. Dynamic recruitment of functionally distinct SWI/SNF chromatin remodeling complexes modulates PDX1 activity in islet β cells. Cell Rep. 2015, 10, 2032–2042. [Google Scholar] [CrossRef] [PubMed]
- Khursheed, M.; Kolla, J.N.; Kotapalli, V.; Gupta, N.; Gowrishankar, S.; Uppin, S.G.; Sastry, R.A.; Koganti, S.; Sundaram, C.; Pollack, J.R.; et al. ARID1B, a member of the human SWI/SNF chromatin remodeling complex, exhibits tumour-suppressor activities in pancreatic cancer cell lines. Br. J. Cancer 2013, 108, 2056–2062. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. BRG1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.; Hamilton, A.J.; Voinnet, O.; Thomas, C.L.; Maule, A.J.; Baulcombe, D.C. RNA–DNA interactions and DNA methylation in post-transcriptional gene silencing. Plant Cell 1999, 11, 2291–2301. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Bartel, D.P. Small RNAs correspond to centromere heterochromatic repeats. Science 2002, 297. [Google Scholar] [CrossRef] [PubMed]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Bartolomei, M.S. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 2013, 152, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.K.; Kuroda, M.I. Noncoding RNAs and intranuclear positioning in monoallelic gene expression. Cell 2007, 128, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Lynn, F.C.; Skewes-Cox, P.; Kosaka, Y.; McManus, M.T.; Harfe, B.D.; German, M.S. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes 2007, 56, 2938–2945. [Google Scholar] [CrossRef] [PubMed]
- Prevot, P.P.; Augereau, C.; Simion, A.; van den Steen, G.; Dauguet, N.; Lemaigre, F.P.; Jacquemin, P. Let-7b and miR-495 stimulate differentiation and prevent metaplasia of pancreatic acinar cells by repressing HNF6. Gastroenterology 2013, 145, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, L.; An, Y.; Zhang, Z.W.; Lang, Y.; Tai, S.; Guo, F.; Teng, C.B. miR-18a regulates expression of the pancreatic transcription factor PTF1A in pancreatic progenitor and acinar cells. FEBS Lett. 2012, 586, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Jin, L.; Wang, X.; Luo, A.; Hu, J.; Zheng, X.; Tsark, W.M.; Riggs, A.D.; Ku, H.T.; Huang, W. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, 17892–17897. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, W.P.; Lagendijk, A.K.; Ketting, R.F.; Moulton, J.D.; Plasterk, R.H. Targeted inhibition of miRNA maturation with morpholinos reveals a role for miR-375 in pancreatic islet development. PLoS Biol. 2007, 5, e203. [Google Scholar] [CrossRef] [PubMed]
- Kredo-Russo, S.; Mandelbaum, A.D.; Ness, A.; Alon, I.; Lennox, K.A.; Behlke, M.A.; Hornstein, E. Pancreas-enriched miRNA refines endocrine cell differentiation. Development 2012, 139, 3021–3031. [Google Scholar] [CrossRef] [PubMed]
- Caponi, S.; Funel, N.; Frampton, A.E.; Mosca, F.; Santarpia, L.; van der Velde, A.G.; Jiao, L.R.; de Lio, N.; Falcone, A.; Kazemier, G.; et al. The good, the bad and the ugly: A tale of miR-101, miR-21 and miR-155 in pancreatic intraductal papillary mucinous neoplasms. Ann. Oncol. 2013, 24, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, O.; Takamori, H.; Iwatsuki, M.; Baba, Y.; Sakamoto, Y.; Tanaka, H.; Chikamoto, A.; Horino, K.; Beppu, T.; Kanemitsu, K.; et al. Carcinogenesis of intraductal papillary mucinous neoplasm of the pancreas: Loss of microRNA-101 promotes overexpression of histone methyltransferase EZH2. Ann. Surg. Oncol. 2012, 19, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Helm, J.; Coppola, D.; Kim, D.; Malafa, M.; Kim, S.J. MicroRNAs in pancreatic ductal adenocarcinoma. World J. Gastroenterol. 2011, 17, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of mir-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hao, J.; Xie, F.; Hu, X.; Liu, C.; Tong, J.; Zhou, J.; Wu, J.; Shao, C. Downregulation of miR-132 by promoter methylation contributes to pancreatic cancer development. Carcinogenesis 2011, 32, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Li, Z.; Wang, X.; Xu, P.; Zhao, L.; Qian, J. miR-125a regulates chemo-sensitivity to gemcitabine in human pancreatic cancer cells through targeting A20. Acta Biochim. Biophys. Sin. 2016, 48, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Habbe, N.; Koorstra, J.B.; Mendell, J.T.; Offerhaus, G.J.; Ryu, J.K.; Feldmann, G.; Mullendore, M.E.; Goggins, M.G.; Hong, S.M.; Maitra, A. MicroRNA miR-155 is a biomarker of early pancreatic neoplasia. Cancer Boil. Ther. 2009, 8, 340–346. [Google Scholar] [CrossRef]
- Farrell, J.J.; Toste, P.; Wu, N.; Li, L.; Wong, J.; Malkhassian, D.; Tran, L.M.; Wu, X.; Li, X.; Dawson, D.; et al. Endoscopically acquired pancreatic cyst fluid microRNA 21 and 221 are associated with invasive cancer. Am. J. Gastroenterol. 2013, 108, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Yuan, Y.; Zhang, C.; Zhang, C.; Yao, W.; Wang, C.; Liu, R.; Ba, Y. Identification of circulating miR-25 as a potential biomarker for pancreatic cancer diagnosis. Cell. Physiol. Biochem. 2016, 39, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- Hussein, N.A.; Kholy, Z.A.; Anwar, M.M.; Ahmad, M.A.; Ahmad, S.M. Plasma miR-22-3p, miR-642b-3p and miR-885-5p as diagnostic biomarkers for pancreatic cancer. J. Cancer Res. Clin. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Chang, P.; LeBlanc, A.; Li, D.; Abbruzzesse, J.L.; Frazier, M.L.; Killary, A.M.; Sen, S. Micrornas in plasma of pancreatic ductal adenocarcinoma patients as novel blood-based biomarkers of disease. Cancer Prev. Res. 2009, 2, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Morimura, R.; Komatsu, S.; Ichikawa, D.; Takeshita, H.; Tsujiura, M.; Nagata, H.; Konishi, H.; Shiozaki, A.; Ikoma, H.; Okamoto, K.; et al. Novel diagnostic value of circulating miR-18a in plasma of patients with pancreatic cancer. Br. J. Cancer 2011, 105, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Cunningham, D.; Peckitt, C.; Barton, S.; Tait, D.; Hawkins, M.; Watkins, D.; Starling, N.; Rao, S.; Begum, R.; et al. miR-21 expression and clinical outcome in locally advanced pancreatic cancer: Exploratory analysis of the pancreatic cancer ERBITUX, radiotherapy and UFT (PERU) trial. Oncotarget 2016, 7, 12672–12681. [Google Scholar] [PubMed]
- Kawaguchi, T.; Komatsu, S.; Ichikawa, D.; Morimura, R.; Tsujiura, M.; Konishi, H.; Takeshita, H.; Nagata, H.; Arita, T.; Hirajima, S.; et al. Clinical impact of circulating miR-221 in plasma of patients with pancreatic cancer. Br. J. Cancer 2013, 108, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Du, Y.; Wang, G.; Gao, J.; Gong, Y.; Li, L.; Zhang, Z.; Zhu, J.; Jing, Q.; Qin, Y.; et al. Detection of differentially expressed microRNAs in serum of pancreatic ductal adenocarcinoma patients: miR-196a could be a potential marker for poor prognosis. Dig. Dis. Sci. 2011, 56, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Passadouro, M.; Faneca, H. Managing pancreatic adenocarcinoma: A special focus in microRNA gene therapy. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G. miR-34—A microRNA replacement therapy is headed to the clinic. Front. Genet. 2012, 3, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Badalian-Very, G. Clinical applications of microRNAs. F1000Research 2013, 2, 136–152. [Google Scholar] [CrossRef] [PubMed]
- Ku, G.M.; Kim, H.; Vaughn, I.W.; Hangauer, M.J.; Myung Oh, C.; German, M.S.; McManus, M.T. Research resource: RNA-SEQ reveals unique features of the pancreatic β cell transcriptome. Mol. Endocrinol. 2012, 26, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Moran, I.; Akerman, I.; van de Bunt, M.; Xie, R.; Benazra, M.; Nammo, T.; Arnes, L.; Nakic, N.; Garcia-Hurtado, J.; Rodriguez-Segui, S.; et al. Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012, 16, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Pullen, T.J.; Rutter, G.A. Roles of lncRNAs in pancreatic β cell identity and diabetes susceptibility. Front. Genet. 2014, 5, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Hu, H.; Zhuo, M.; Wang, L.; Cui, J.J.; Jiao, F.; Wang, L.W. Long non-coding RNA: An emerging paradigm of pancreatic cancer. Curr. Mol. Med. 2016, 16, 702–709. [Google Scholar] [CrossRef]
- Zheng, J.; Huang, X.; Tan, W.; Yu, D.; Du, Z.; Chang, J.; Wei, L.; Han, Y.; Wang, C.; Che, X.; et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat. Genet. 2016, 48, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Nong, K.; Zhu, H.; Wang, W.; Huang, X.; Yuan, Z.; Ai, K. H19 promotes pancreatic cancer metastasis by derepressing let-7’s suppression on its target HMGA2-mediated EMT. Tumour. Biol. 2014, 35, 9163–9169. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tian, X.; Wang, F.; Zhang, Z.; Du, C.; Xie, X.; Kornmann, M.; Yang, Y. The long noncoding RNA H19 promotes cell proliferation via E2F-1 in pancreatic ductal adenocarcinoma. Cancer Boil. Ther. 2016, 17, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, G.Q.; Chen, H.; Zhao, Z.J.; Chen, H.Z.; Liu, H.; Wang, G.; Jia, Y.H.; Pan, S.H.; Kong, R.; et al. Plasma and tumor levels of linc-pint are diagnostic and prognostic biomarkers for pancreatic cancer. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, J.; Yuan, X.; Qian, W.; Zhang, B.; Shi, M.; Xie, J.; Shen, B.; Xu, H.; Hou, Z.; et al. Long noncoding RNA uc.345 promotes tumorigenesis of pancreatic cancer by upregulation of hnRNPL expression. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Chen, X.; Li, J.; Guo, Y.; Li, H.; Pan, X.; Jiang, J.; Liu, H.; Wu, B. Salivary HOTAIR and PVT1 as novel biomarkers for early pancreatic cancer. Oncotarget 2016, 7, 25408–25419. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Ohana, P.; Konikoff, F.M.; Leichtmann, G.; Hubert, A.; Appelbaum, L.; Kopelman, Y.; Czerniak, A.; Hochberg, A. Phase 1/2A, dose-escalation, safety, pharmacokinetic and preliminary efficacy study of intratumoral administration of BC-819 in patients with unresectable pancreatic cancer. Cancer Gene Ther. 2012, 19, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, A.A.; Chen, Y.B.; Wren, J.; Gonen, M.; Abdel-Wahab, O.; Heguy, A.; Liu, H.; Takeda, S.; Tickoo, S.K.; Reuter, V.E.; et al. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur. Urol. 2013, 63, 848–854. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Epigenetic Mechanism | Protein/RNA | Alterations in PDA |
|---|---|---|
| DNA methylation | DNMT1, DNMT3A, DNMT3B | Increased expression |
| Histone modification | ||
| Histone methylation | KDM6A | Loss of copy number, single nucleotide variation, deletion, amplification |
| MLL2 | Loss of copy number, single nucleotide variation, deletion | |
| EZH2 | Increased expression | |
| G9a | Increased expression | |
| Histone acetylation | p300 | Decreased expression, missense mutation |
| HDAC1-3, HDAC7 | Increased expression | |
| Histone ubiquitilation | H2AK119Ub1, Ring2, Bim1 | Increased expression |
| Chromatin remodeling | ARID1A, ARID1B, PBRM1, SMARCA2, SMARCA4, ARID2, BRD7 | Mutations |
| SMARCA2 | Promoter polymorphyisms, increased expression | |
| BAF180 | Decreased expression | |
| Non coding RNA | ||
| Small RNAs | miR-21, miR-155, miR-196a-2, miR-203, miR-210, miR-222, miR-25, miR-22-3p, miR-642b-3p, miR-885-5p, miR-18a | Increased expression |
| miR-101, miR-34a | Decreased expression | |
| lncRNAs | LINC00673 | Germline mutation |
| H19, uc.345, HOTAIR, HOTTIP, PVT1 | Increased expression | |
| Linc-pint | Decreased expression | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silverman, B.R.; Shi, J. Alterations of Epigenetic Regulators in Pancreatic Cancer and Their Clinical Implications. Int. J. Mol. Sci. 2016, 17, 2138. https://doi.org/10.3390/ijms17122138
Silverman BR, Shi J. Alterations of Epigenetic Regulators in Pancreatic Cancer and Their Clinical Implications. International Journal of Molecular Sciences. 2016; 17(12):2138. https://doi.org/10.3390/ijms17122138
Chicago/Turabian StyleSilverman, Brittany R., and Jiaqi Shi. 2016. "Alterations of Epigenetic Regulators in Pancreatic Cancer and Their Clinical Implications" International Journal of Molecular Sciences 17, no. 12: 2138. https://doi.org/10.3390/ijms17122138
APA StyleSilverman, B. R., & Shi, J. (2016). Alterations of Epigenetic Regulators in Pancreatic Cancer and Their Clinical Implications. International Journal of Molecular Sciences, 17(12), 2138. https://doi.org/10.3390/ijms17122138