Genomic and Clinical Correlates of Adrenocortical Carcinoma in an Adult Patient with Li-Fraumeni Syndrome: A Case Report


 ,
,
Abstract
:1. Introduction
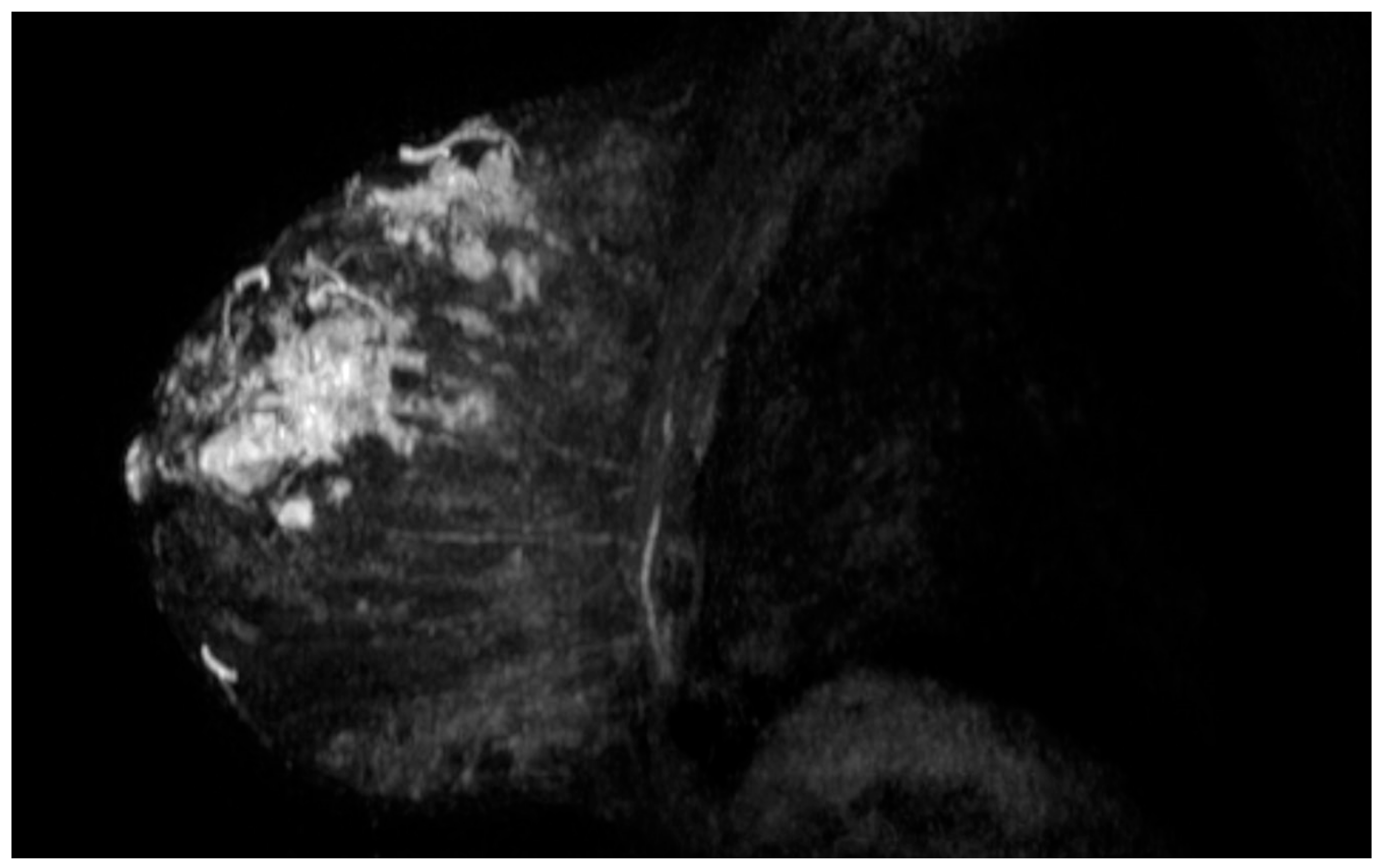
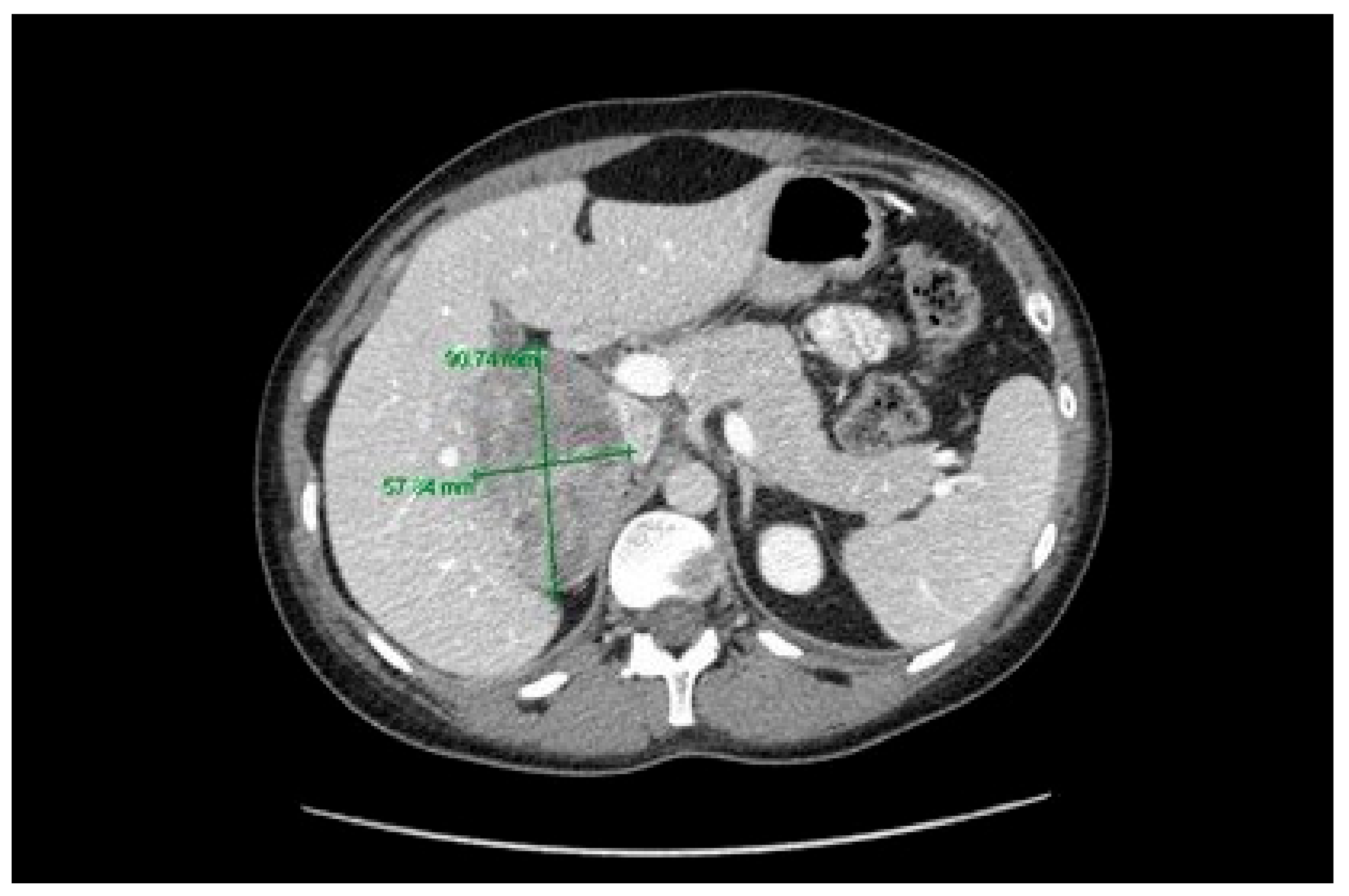
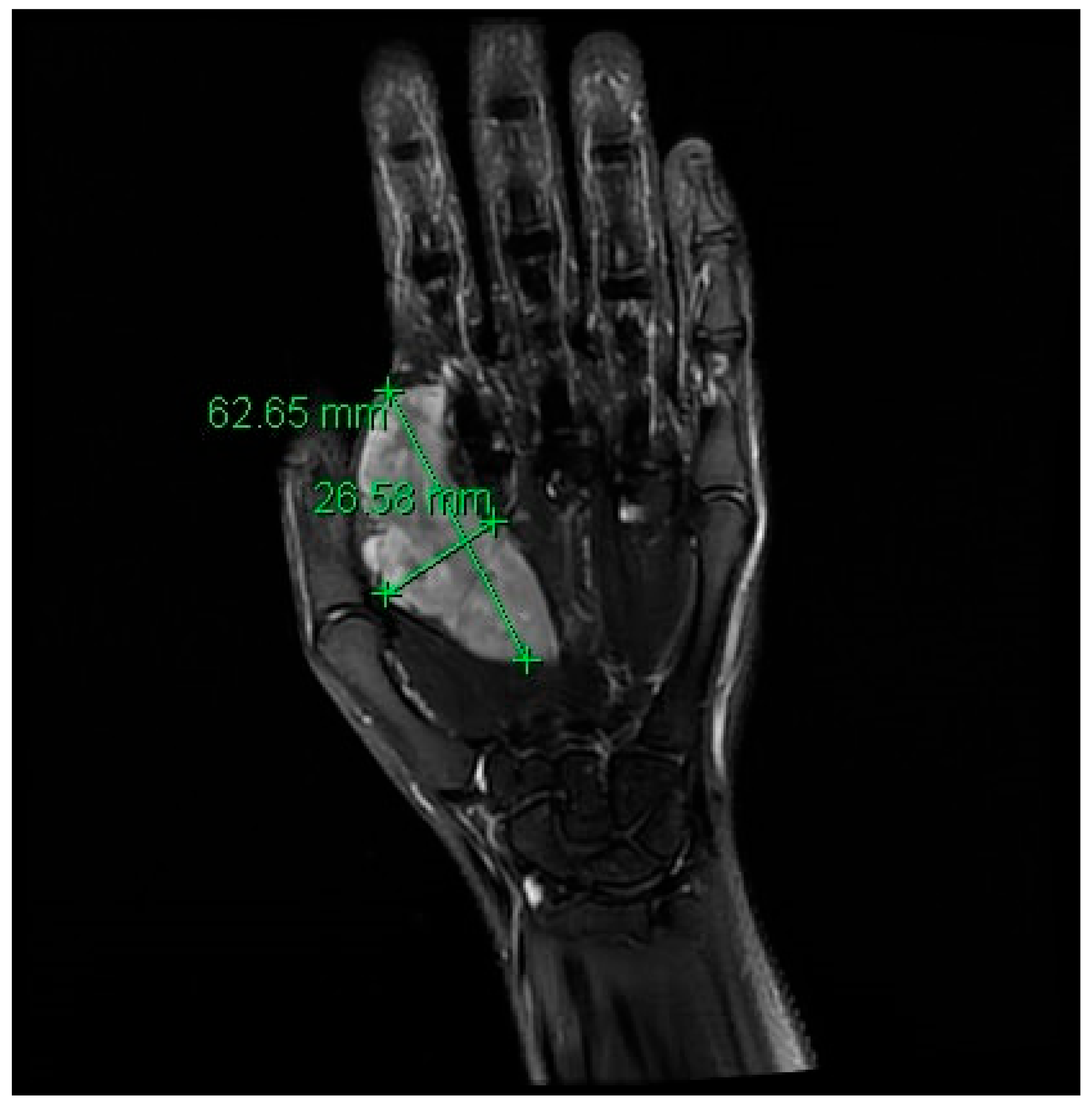
2. Case Description
3. Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Malkin, D. Li-Fraumeni Syndrome. Genes Cancer 2011, 2, 475–484. [Google Scholar]
- Schneider, K.; Zelley, K.; Nichols, K.E.; Garber, J. Li-Fraumeni Syndrome. GeneReviews. 1999. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1311/ (accessed on 30 December 2020).
- Wasserman, J.D.; Zambetti, G.P.; Malkin, D. Towards an understanding of the role of p53 in adrenocortical carcinogenesis. Mol. Cell. Endocrinol. 2012, 351, 101–110. [Google Scholar]
- Herrmann, L.J.; Heinze, B.; Fassnacht, M.; Willenberg, H.S.; Quinkler, M.; Reisch, N.; Zink, M.; Allolio, B.; Hahneret, S. TP53 germline mutations in adult patients with adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, E476–E485. [Google Scholar]
- FoundationOne CDx. Technical Information; Foundation Medicine: Cambridge, UK, 2019; p. 1. Available online: https://assets.ctfassets.net/vhribv12lmne/6Rt6csmCPuaguuqmgi2iY8/2fe839f0e9075cf4a047bf241374e6af/F1CDx.Label.Technical_Info_Final_July_2019.pdf (accessed on 26 November 2019).
- Ayala-Ramirez, M.; Jasim, S.; Feng, L.; Ejaz, S.; Deniz, F.; Busaidy, N.; Waguespack, S.G.; Naing, A.; Sircar, K.; Wood, C.G.; et al. Adrenocortical carcinoma: Clinical outcomes and prognosis of 330 patients at a tertiary care center. Eur. J. Endocrinol. 2013, 169, 891. [Google Scholar]
- Luton, J.P.; Cerdas, S.; Billaud, L.; Thomas, G.; Guilhaume, B.; Bertagna, X.; Laudat, M.H.; Louvel, A.; Chapuis, Y.; Blondeau, P.; et al. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N. Engl. J. Med. 1990, 322, 1195–1201. [Google Scholar]
- Fassnacht, M.; Libé, R.; Kroiss, M.; Allolio, B. Adrenocortical carcinoma: A clinician’s update. Nat. Rev. Endocrinol. 2011, 7, 323–335. [Google Scholar]
- Fassnacht, M.; Dekkers, O.M.; Else, T.; Baudin, E.; Berruti, A.; De Krijger, R.R.; Haak, H.; Mihai, R.; Assie, G.; Terzolo, M. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2018, 179, G1–G46. [Google Scholar]
- Fay, A.P.; Signoretti, S.; Callea, M.; Telό, G.H.; McKay, R.R.; Song, J.; Carvo, I.; Lampron, M.E.; Kaymakcalan, M.D.; Poli-de-Figueiredo, C.E.; et al. Programmed death ligand-1 expression in adrenocortical carcinoma: An exploratory biomarker study. J. Immunother. Cancer 2015, 3, 3. [Google Scholar]
- Sorrell, A.D.; Espenschied, C.R.; Culver, J.O.; Weitzel, J.N. Tumor protein p53 (TP53) testing and Li-Fraumeni syndrome: Current Status of Clinical Applications and Future Directions. Mol. Diagn. Ther. 2013, 17, 31–47. [Google Scholar]
- Assié, G.; Letouzé, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; René-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar]
- Wang, W.; Han, R.; Ye, L.; Xie, J.; Tao, B.; Sun, F.; Zhuo, R.; Chen, X.; Den, X.; Ye, C.; et al. Adrenocortical carcinoma in patients with MEN1: A kindred report and review of the literature. Endocr. Connect. 2019, 8, 230–238. [Google Scholar]
- Shimura, T.; Tada, Y.; Hirai, H.; Kawakita, D.; Kano, S.; Tsukahara, K.; Shimizu, A.; Takase, S.; Imanishi, Y.; Ozawa, H.; et al. Prognostic and histogenetic roles of gene alteration and the expression of key potentially actionable targets in salivary duct carcinomas. Oncotarget 2018, 9, 1852. [Google Scholar]
- Fiorentini, C.; Fragni, M.; Tiberio, G.A.; Galli, D.; Roca, E.; Salvi, V.; Bosisio, D.; Missale, C.; Terzolo, M.; Memo, M.; et al. Palbociclib inhibits proliferation of human adrenocortical tumor cells. Endocrine 2018, 59, 213–217. [Google Scholar]
- Assi, T.; Kattan, J.; Rassy, E.; Nassereddine, H.; Farhat, F.; Honore, C.; Cesne, A.L.; Adam, J.; Mir, O. Targeting CDK4 (cyclin-dependent kinase) amplification in liposarcoma: A comprehensive review. Crit. Rev. Oncol. Hematol. 2020, 153, 103029. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Tissue Sample | ERBB2 | FGFR4 | NOTCH3 | BRCA1 | BRCA2 | MEN1 | TP53 |
|---|---|---|---|---|---|---|---|
| Breast invasive ductal carcinoma | amplification | amplification | amplification | negative | negative | P191fs*18 | |
| Adrenocortical Carcinoma | Splice site 840—2A>T | P191fs*18 | |||||
| Soft Tissue Sarcoma | P191fs*18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondy, S.; Tajzler, C.; Hotte, S.J.; Kapoor, A.; Zbuk, K.; Lalani, A.-K.A. Genomic and Clinical Correlates of Adrenocortical Carcinoma in an Adult Patient with Li-Fraumeni Syndrome: A Case Report. Curr. Oncol. 2021, 28, 226-232. https://doi.org/10.3390/curroncol28010025
Bondy S, Tajzler C, Hotte SJ, Kapoor A, Zbuk K, Lalani A-KA. Genomic and Clinical Correlates of Adrenocortical Carcinoma in an Adult Patient with Li-Fraumeni Syndrome: A Case Report. Current Oncology. 2021; 28(1):226-232. https://doi.org/10.3390/curroncol28010025
Chicago/Turabian StyleBondy, Suraya, Camilla Tajzler, Sebastien J. Hotte, Anil Kapoor, Kevin Zbuk, and Aly-Khan A. Lalani. 2021. "Genomic and Clinical Correlates of Adrenocortical Carcinoma in an Adult Patient with Li-Fraumeni Syndrome: A Case Report" Current Oncology 28, no. 1: 226-232. https://doi.org/10.3390/curroncol28010025
APA StyleBondy, S., Tajzler, C., Hotte, S. J., Kapoor, A., Zbuk, K., & Lalani, A.-K. A. (2021). Genomic and Clinical Correlates of Adrenocortical Carcinoma in an Adult Patient with Li-Fraumeni Syndrome: A Case Report. Current Oncology, 28(1), 226-232. https://doi.org/10.3390/curroncol28010025