Abstract
Background: TP53 is frequently mutated in solid tumors, but its basic mutation mapping is mixed, particularly in aggressive-stage lung cancer. Experimental Design: We curated a total of 139 advanced non-small cell lung cancer (NSCLC) patients who harbored wild-type TP53 (TP53wt) or mutated TP53 (TP53mut) based on next-generation sequencing (NGS) to analyze multiple-dimensional data types, including tumor mutation burden (TMB), programmed death receptor ligand 1 (PD-L1) expression, co-mutant alterations, hotspot mutations distribution, and therapy response. Results: TP53 was evident in 125 mutations and significantly associated with male sex, adenocarcinoma differentiation, smoking history, PD-L1 tumor proportion score, and TMB level. The most frequent mutations were distributed on exon 8, but there were no distinct hotspot mutations. After outlining the co-mutation genes, it is interesting to note that DNA damage repair (DDR) genes were frequent alterations in the mutated TP53 cohort. Even though there was no significant difference between the TP53wt and TP53mut cohorts on therapy response, patients with nucleotide variation in G>T achieved a relatively higher durable clinical benefit (DCB) rate. Conclusions: This real-world retrospective study suggests that molecular stratification on the basis of TP53 mutations should be deeply explored for NSCLC to optimize and modify clinical therapy choices.
1. Introduction
Lung cancer is one of the leading causes of cancer-related mortality worldwide, with a poor 5-year survival rate of 19% [1]. There are two main forms of lung cancer: small cell lung cancer (SCLC, ~15% of patients) and non-small cell lung cancer (NSCLC, ~85% of patients), with the latter further subdivided into two main types: lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LSCC) [2]. Nowadays, an in-depth understanding of genetic alteration and molecular profiling based on next-generation sequencing (NGS) has dramatically changed the treatment landscape of lung cancer to precision therapy.
The tumor suppressor p53 (TP53) gene encodes p53 protein, which consists of 393 amino acids, with four major functional domains: transcriptional, DNA binding, tetramerization, and regulatory domains [3]. Mutations in p53 occur in approximately half of the solid tumors, resulting in loss of tumor suppressor function (impaired expression) or gain of oncogenic activity (aberrant expression). Other than DNA damage response, the exact role of p53 in tumor suppression has been attributed to intracellular metabolism, genetic and epigenetic stability, inflammation remodeling, and integration with various pathways [4,5]. It is observed that amino acid locations at 175, 245, 248, 249, 273, and 282 are frequent hotspot mutations in most cancers and mutant p53 activity may alter tumor therapy response [6,7]. Hence, the understanding of the relationship between p53 modification and tumor inflammatory environment may contribute to more effective cancer treatment. This real-world retrospective cohort study included patients with advanced NSCLC and focused on the roles of both wild-type and mutant forms of p53 in the regulation of tumor development, such as tumor mutational burden (TMB), the level of programmed death receptor ligand 1 (PD-L1) protein on the surface of tumor tissue, their interplay with other genetic alterations and therapy response.
2. Materials and Methods
2.1. Patients and Eligibility
The study retrospectively reviewed 287 patients with advanced lung cancer at our Oncology Department between December 2019 and January 2022. The exclusion criteria were as follows: lost to follow-up (n = 31), refused tissue-based next-generation sequencing (NGS) (n = 59), failed fine-needle aspiration (FNA) biopsy for routinely screened of molecular alterations (n = 26), absence of detailed NGS information (n = 21), and evaluating mutational status with blood-cell-free DNA (cfDNA) (n = 11). Finally, a total of 109 patients with wild-type TP53 or mutant TP53 reported on NGS panels met the inclusion criteria in the present study (Figure 1). The study was reviewed and approved by the Ethics Committee of our department.
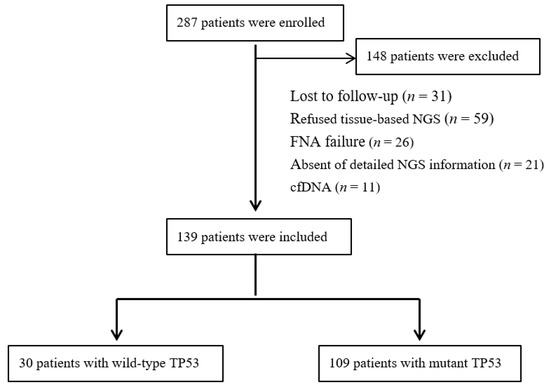
Figure 1.
Flowchart of patient selection.
2.2. Patient Variables
The variables collected included patient demographics (such as age, sex, and smoking history), histological type (lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LSCC), or sarcomatoid carcinoma), programmed death receptor ligand 1 (PD-L1), tumor mutational burden (TMB), and molecular alteration status. Durable clinical benefit (DCB) was defined as a partial response/stable disease that lasted more than 6 months, while no durable benefit (NDB) was defined as a stable disease that lasted less than 6 months. Patient outcomes were characterized by RECIST V.1.1 criteria in combination with clinical notes.
2.3. Tissue-Based Next-Generation Sequencing
DNA extracted from formalin-fixed tumor samples was analyzed with a comprehensive genomic profiling assay that targeted all exons of 825 cancer-related genes.
TMB: Panel_var_filter software (v1.0) was employed to filter variant annotation results, including specific types of variants (non-coding region variants, synonymous mutations), mutation frequency, reads support, variants exclude the coding region of panel 825 genes, and driver gene variants. Finally, the number of filtered variants was calculated to evaluate the TMB value.
PD-L1: Immunohistochemical technique was employed to calculate CPS and TPS values.
2.4. TP53 Mutation Classification
Traditionally, there are two classification systems for TP53 analysis: mutational type and“Poeta rules” [7,8]. The former analyzed the “technical” type of TP53 mutation, while Poeta et al. revealed a classification of the missense mutation group into “disruptive” and “nondisruptive” mutations. Mutant p53 can be also divided into two subtypes, DNA contact and conformational (misfolded/unfolded) mutants [9]. Here, we employed a dichotomous classification as “wild-type” and “mutant” to analyze TP53 activity and further separated the missense mutation (termed “TP53 missense mutations”) from all other mutations, including deletion, synonymous, nonsense, insertion, and frame-shift mutations (termed “TP53other mutations”) based on next-generation sequencing.
2.5. Statistical Analysis
Statistical analyses were conducted using GraphPad Prism (version 9.4.0). The association of the qualitative variables was tested for by chi-square or Fisher’s exact test, depending on distributional assumptions. Scatter dot plots indicate median and 95% confidence interval (CI). All tests were two-sided, and p-values of <0.05 indicated statistical significance.
3. Results
3.1. Patient Outcomes
Table 1 summarized the demographics and characteristics of the patients with advanced lung cancer. The follow-up period ranged from 189 to 757 days, with a median follow-up time for censored patients of 414 days. A total of 109 patients (78.4%) carried TP53 mutations. Most of the patients were of the male sex (73.4%) and had adenocarcinoma (59.7%). More than half of the patients had a smoking history (72.7%). Mutations in TP53 were significantly associated with male sex, adenocarcinoma differentiation, smoking history, PD-L1 tumor proportion score, and TMB level.

Table 1.
Demographics and characteristics for patients with TP53wt and TP53mut in advanced lung cancer.
3.2. Association with PD-L1 and TMB Expression
Furthermore, scatter plots for PD-L1 tumor proportion score and TMB level are shown in Figure 2. There were 30 patients with TP53wt who revealed a relatively lower expression of PD-L1 and TMB compared with 109 patients in the TP53mut cohort.
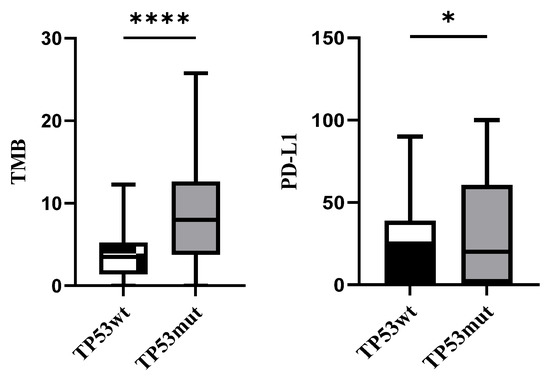
Figure 2.
TMB and PD-L1 levels in TP53wt and TP53mut cohorts (* p < 0.1, **** p < 0.0001).
3.3. Mutation Mapping for TP53
TP53 was evident in 125 mutations, in which the majority were missense mutations (82), followed by nonsense (18), frame-shift (15), and other mutations, including insertion and deletion mutations (Figure 3A). More than half of TP53 mutations were clustered in the DNA binding domain (95), followed by tetramerization, proline-rich, regulatory, and transactivation domains (Figure 3B). The most frequent missense point mutations were distributed on exon 8(30), followed by exon 5(29), 7(28), 4(15), 9(8), 10(8), and 6(7), respectively. Accordingly, mutp53 can be divided into two subtypes, DNA contact (codons 248 and 273) and conformational (codons 157, 158, 179, 245, and 249) mutants. Here, we identified the most frequent hotspots being codons 273(7), 158(6), and 175(4). There were no distinct hotspot mutations (Figure 3C).
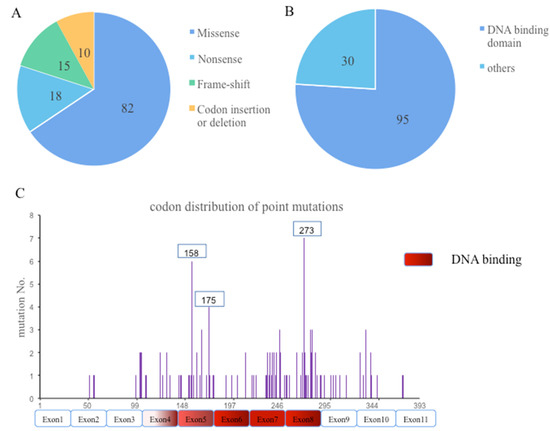
Figure 3.
“Technical” types (A), cluster of domains (B), and codon distribution (C) of TP53 mutation in 109 patients.
3.4. Co-Mutation Status in TP53wt and TP53mut Cohort
A series of studies demonstrate that P53 is a central tumor suppressor and the TP53 mutations display substantial immune cell composition and increased immune response [10]. Research on the co-occurring of TP53 and different mutations could help to translate the molecular findings into clinical decision-making. Here, we employed beneficial and noxious genes related to cancer treatment to evaluate their correlation with TP53wt and TP53mut. For mutated TP53, clinically relevant commutations were distributed as follows: 22.9% had EGFR mutations, 15.6% KRAS mutations, 15.6% KMT2D mutations, 12.8% ARID1A mutations, 12.8% KEAP1 mutations, and 61.5% with DDR gene mutations (Table 2).
Table 2.
Co-mutational status for patients with TP53wt and TP53mut in advanced lung cancer.
3.5. Therapy Response for Patients with TP53wt and TP53mut
NSCLC with TP53 mutations had a 56.0% rate of DCB, which revealed no significant difference with the wild-type cohort (Figure 4). Out of 17 patients in the TP53wt group, 3 who underwent immunotherapy were diagnosed with an adverse immune-related disease, compared with 9 out of 73 patients in the mutTP53 cohort. Significantly, patients with EGFR mutation had a lower DCB rate compared with the TP53 mutation cohort (p < 0.01). Furthermore, for TP53 missense mutation, nucleotide variation in G>T was achieved at a relatively higher DCB rate.
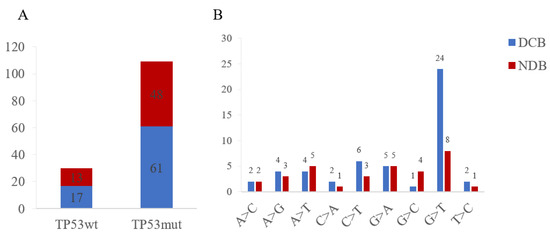
Figure 4.
Clinical benefit (A) and nucleotide variation (B) of TP53wt and TP53mut cohorts. DCB, durable clinical benefit; NDB, no durable benefit.
4. Discussion
Emerging evidence describes the regulatory mechanism of p53 in the metabolism balance and inflammatory response. It is recognized that LSCC frequently carries TP53 mutations related to carcinogen exposure and most p53 mutants are post-translationally modified at the same residues as wild-type status [11,12]. Hence, we summarized the demographics and characteristics of the censored 139 patients with advanced-stage lung cancer and found that more than half of the patients were of male sex and had a smoking history. Even though there was no obvious difference between TP53wt and TP53mut status in baseline characteristics, the TP53 mutant cohort revealed a significant association with male sex, adenocarcinoma differentiation, and smoking history.
TMB represents the overall number of mutations per megabase of DNA and is consequentially related to the number of neoepitopes that subsequently trigger T cell response. PD-L1 is a transmembrane cell surface protein, which is expressed on the tumor cells and inhibits T cell immune response [13]. Accordingly, TMB has been shown to be a potential predictor of response to immunotherapy across various tumor types, and increased TMB levels correlate with several factors, such as tobacco exposure and defective tumor suppressor gene TP53 [14]. In addition to PD-L1 expression level, our study revealed that the TP53mut cohort was much more obviously associated with TMB level, which may be related to lung cancer type and DNA replication errors mediation [15]. Furthermore, we found an inconsistency in the association between PD-L1 expression and TMB level, which is not unexpected (Figure S1).
Here, the majority of TP53 are missense mutations with amino acid changes mainly in the DNA binding domain (residues 102-292). The missense-mutant TP53 acquires gain-of-function (GOF) activities and can be broadly classified as DNA contact mutants and structural mutants, subsequently inactivating other tumor suppressive proteins and promoting tumor progression. Different from traditional hotspot mutation points [9], we identified that mutations in TP53 were scattered and the most frequent hotspots being codons 273(7/125), 158(6/125), and 175(4/125) in advanced lung cancer. Considerable evidence has solidified that the oncogenic function of mutp53 is not only caused by altered structure and properties of GOF, but also by losing its tumor suppressive activity. Thus, a smaller but considerable fraction of TP53 mutations is truncating mutations that impair the wild-type effect and complete loss-of-function (LOF) during tumor development [16].
Although mutations in the p53 gene exist in around half of all human cancers, detailed co-mutation analysis in the real world of advanced lung cancer is limited. Traditionally, p53 pathway activation controls the cellular response to different stress stimuli and determines cell fate under a specific context. Amplification of Mdm2/4 appears to promote tumor cell survival, drug resistance, invasion, and metastasis via E3 ubiquitin-mediated proteasome degradation. Ataxia telangiectasia mutated (ATM), ataxia telangiectasia, and Rad3-related protein (ATR) and the checkpoint kinases CHK1 and CHK2 stabilize p53 by disrupting p53-MDM2 interaction [17]. Numerous mutated genes correlated with tumor therapy response, such as EGFR, KRAS, ARID1A, and SMARCA4. Interestingly, we outlined the co-mutational status of these beneficial and noxious genes with TP53 in lung cancer and found a significantly different status of co-mutated DNA damage repair genes was detected in the TP53mut cohort, which may be related to better immune checkpoint inhibitor response and prognosis.
All patients with TP53wt and TP53mut showed no difference in durable clinical benefit, which may be due to limited samples and the short follow-up period of the analyzed data. Furthermore, a minimal degree of informative censoring was ruled out, leading to an underestimation of survival times. We revealed that 3 out of 17 patients in the TP53wt group were diagnosed with an adverse immune-related disease, compared with 9 out of 73 patients in the mutTP53 cohort who underwent immunotherapy. Thus, the relationship between adverse immune-related disease and mutated TP53 still needs further exploration. Furthermore, nucleotide variation in G>T achieved a relatively higher DCB rate, which might be related to oxidative stress and recognized as a predictor for TP53mut patients [18]. Nowadays, increased recognition of p53 has paved the way for various therapies targeting TP53 to restore normal p53 tumor suppressive function [19]. Studies describing artificial compounds that inactivated TP53 mutations provided a conceptual basis for the feasibility of mutant p53 reactivation [20].
5. Conclusions
In conclusion, this real-world retrospective study provides unparalleled statistical power to the properties of TP53wt and TP53mut in advanced-stage NSCLC. TP53 mutant was not only associated with male sex, adenocarcinoma differentiation, and smoking history, but also affected PD-L1 expression, TMB level, and co-mutant alterations. Our data also identified the prognostic value of nucleotide variation in G>T and molecular stratification on the basis of TP53 mutations should be broadened for NSCLC to optimize and modify clinical therapy choices.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/curroncol29100582/s1, Figure S1: The association between TMB and PD-L1 levels in TP53mut cohort.
Author Contributions
Conceptualization, F.H.; Formal analysis, F.H.; Investigation, F.H. and L.G.; Methodology, F.H.; Resources, F.H.; Software, F.H.; Supervision, D.Z.; Visualization, F.H.; Writing—original draft, F.H.; Writing—review and editing, F.H. and D.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was approved by the Ethical Committee of Tianjin Medical University General Hospital (NO.IRB2022-WZ-110, August, 2022).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All data, models, and code generated or used during the study appear in the submitted article.
Acknowledgments
The authors would like to thank the associate editor and reviewers for their helpful remarks.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to The 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef]
- Chen, Y.; Dey, R.; Chen, L. Crystal Structure of the p53 Core Domain Bound to a Full Consensus Site as a Self-Assembled Tetramer. Structure 2010, 18, 246–256. [Google Scholar] [CrossRef]
- Mello, S.S.; Attardi, L.D. Deciphering p53 signaling in tumor suppression. Curr. Opin. Cell Biol. 2018, 51, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hickman, J.H.; Wang, S.-J.; Gu, W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 2015, 14, 2881–2885. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Poeta, M.L.; Manola, J.; Goldwasser, M.A.; Forastiere, A.; Benoit, N.; Califano, J.A.; Ridge, J.A.; Goodwin, J.; Kenady, D.; Saunders, J.; et al. TP53Mutations and Survival in Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2007, 357, 2552–2561. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol. Cell 2018, 71, 873. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 Mutations in Human Cancer: Database Reassessment and Prospects for the Next Decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Wiman, K. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Li, X.X.; Wang, H.L.; Wang, J.; Chen, Y.Y.; Yin, X.B.; Shi, G.Y.; Li, H.; Hu, Z.Q.; Liang, X.W. Emodin enhances cisplatin-induced cytotoxicity in human bladder cancer cells through ROS elevation and MRP1 downregulation. BMC Cancer 2016, 16, 578. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Blanden, A.R.; Yu, X.; Wolfe, A.J.; Gilleran, J.A.; Augeri, D.J.; O’Dell, R.S.; Olson, E.C.; Kimball, S.D.; Emge, T.J.; Movileanu, L.; et al. Synthetic Metallochaperone ZMC1 Rescues Mutant p53 Conformation by Transporting Zinc into Cells as an Ionophore. Mol. Pharmacol. 2015, 87, 825–831. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).