1. Introduction
Extracellular vesicles (EVs) are nano-sized vesicles released by various cell types that have functions of cell-to-cell communication at various distances, maintenance of organ homeostasis, and induction of diseases [
1,
2,
3]. It is expected that the functions of EVs will reflect the properties of the original cells that released those EVs.
A nutritive microenvironment or niche is required for disseminating tumor cells to engraft distant sites. The concept of a niche is based on the ‘seed and soil’ hypothesis [
4]. Pancreatic ductal adenocarcinoma-derived EVs, for instance, may induce formation of a pre-metastatic niche in the liver [
5]. Metastatic niche formation is not limited to distant sites. Prostate cancer-associated EVs modulate the tumor microenvironment to prepare a metastatic niche [
6]. EVs containing ErbB2/CRK derived from bladder cancer can induce pre-metastatic niches in lung, liver, and bone [
7]. Tumor cell-derived EVs are believed to stimulate tumorigenesis via promoting angiogenesis [
8], remodeling of extracellular matrix [
9], secretion of inflammatory molecules, and suppression of immune responses [
10,
11].
By contrast to EVs from tumor cells, EVs of histiocytes, immune cells, or cancer cells with low metastatic potential may suppress cancer metastasis; this latter property may be of value for anti-metastasis therapies using an EV-delivery system. Intravenous administration of EVs isolated from non-metastatic melanoma cells has been shown to suppress the metastasis of melanomas [
12]. Similarly non-cancer cell-derived exosomes, whose stability in blood has been increased by the expression of CD47, are effective in treatments targeting the Kras oncogene in pancreatic cancer [
13]. EVs of dendritic cells can promote a tumor antigen-specific response [
14]. Menstrual mesenchymal stem cells inhibit angiogenesis and the growth of oral squamous cell carcinomas [
15]. MC38, a colorectal adenocarcinoma model cell line, has been genetically modified by overexpressing IL-12 to enhance its natural killer cell activation potency; EVs from this modified MC38 cell line can suppress the growth of subcutaneous tumors in the abdominal cavities of mice [
16].
However, given the diversity of cancer cells and the complex factors associated with their metastasis, we posited that the characteristics of EVs might not always reflect the original cancer cells. Moreover, we wondered if highly aggressive cancer cell-derived EVs could be modified into suppressive EVs by cytokines and immune cells during in vivo circulation. If EVs with such properties are available, their role may be an autovaccine against metastasis [
3].
Cancer vaccines have been intensively studied and properties of tumor antigens are summarized in a recent review [
17]. Vaccine types are mostly peptide, glycopeptide, viral, DNA, and mRNA. Although the number is small, there are also cellular type such as dendritic cells, melanoma cells, colon cancer cells, and pancreatic adenocarcinoma cells. However, there is no report on subcellular particles and extracellular vesicles. It seemed to be due to an idea that those fractions might not contain cancer antigens that should exist on the cell surface. In contrast, our idea is that EVs may provide intracellular molecular information to the immune system of the target tissue rather than cell surface antigen information. Then, instead of whole cell, EVs have been investigated on their possible role as a candidate of autovaccine.
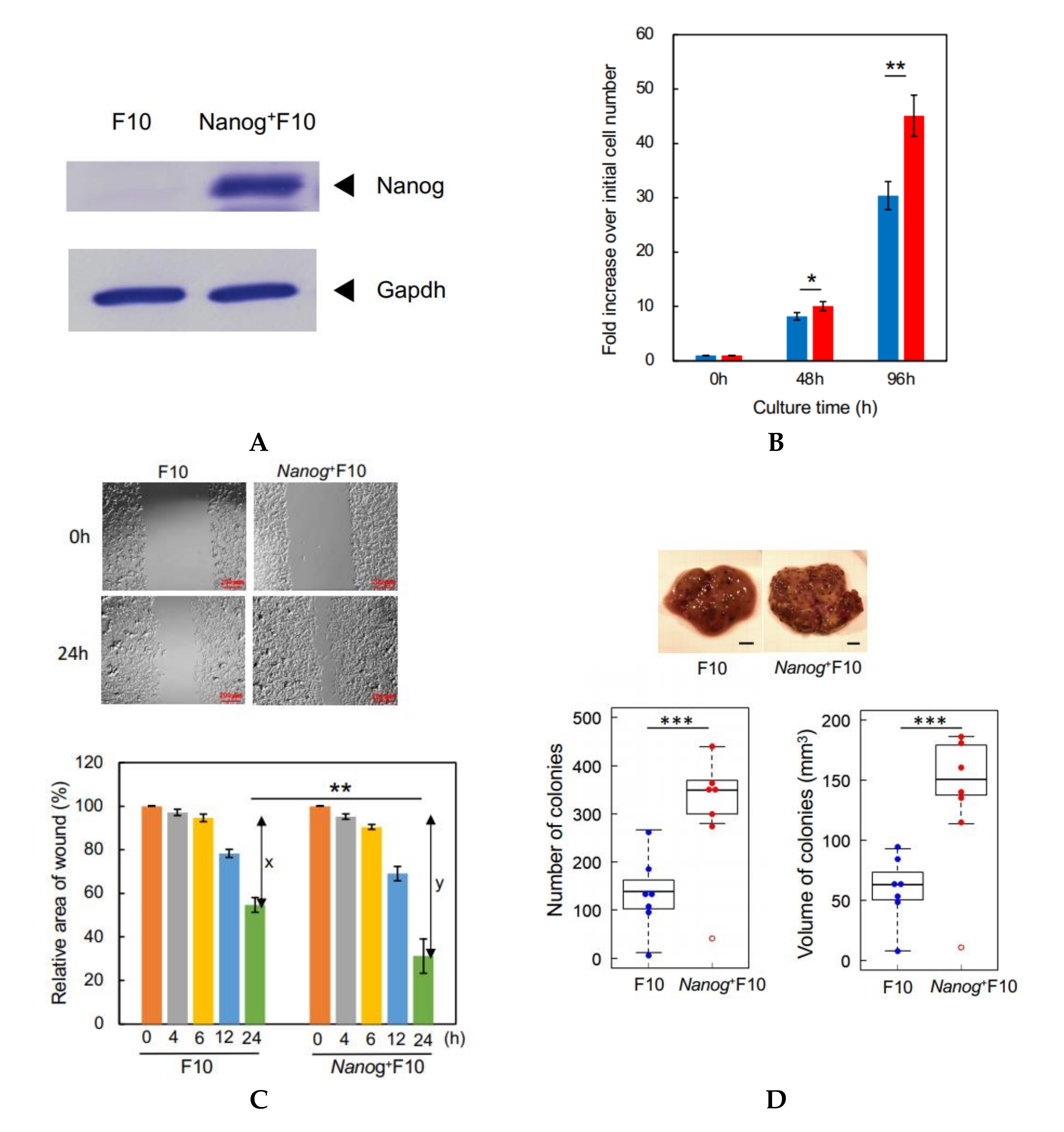
Previously, we modified the melanoma cell line B16-BL6 (BL6) to overexpress
Nanog, a major factor for maintaining the undifferentiated state of embryonic stem cells, in the expectation that this would increase the metastatic potential of this cell line. The modified BL6 line did indeed show an enhanced level of metastatic potential [
18]. A transcriptome sequencing analysis indicated that transforming growth factor (TGF)-β1 was the only secretory cytokine among the top15 up-regulated and top 16 down-regulated genes after
Nanog overexpression in BL6 cells.
Here, we have modified the melanoma cell line F10 to overexpress Nanog. F10 and BL6 are the same strain of melanoma, but with completely different metastatic properties. BL6 does not metastasize to the liver, but F10 most often metastasizes to the liver. Therefore, regarding the effects of Nanog overexpression, although the effect on BL6 was investigated in the previous report, it is necessary to confirm that the same effect on F10 could be obtained as well.
The involvement of TGF-β1 was also assumed in the effects brought about by
Nanog overexpression. TGF-β1 shows a dual role in the regulation of metastasis; tumor-suppressive in early stage tumors, but tumor-promotive in advanced cancer [
19,
20,
21]. Moreover, TGF-β1 can be transferred to distant target cells through EVs [
22]. Therefore, we thought that a key role of TGF-β1 in the suppression of metastasis might be found.
Next, we considered the interaction between TGF-β1 and immune cells. Among immune cells such as macrophages, T-cells, B-cells, and natural killer cells, we focused on macrophages based on the report that TGF-β1 could induce tumor promotive M2-like macrophages [
23]. Immune responses concerning M1- or M2-polarization of macrophages might be related to the metastasis suppression via TGF-β1 signaling.
2. Materials and Methods
2.1. Cell Culture
A mouse melanoma cell line, B16-F10 (F10) cells, and a mouse macrophage cell line, J774.1 cells were cultured in R10 medium (RPMI 1640 containing 10% fetal bovine serum [Thermo Fisher Scientific, Waltham, MA, USA] and penicillin-streptomycin [Thermo Fisher Scientific]) at 37 °C under 5% CO2.
2.2. Animals
This study was carried out in compliance with the ARRIVE guidelines [
24]. C57BL/6 male mice were bred in a specific pathogen-free room under conditions of 12 h illumination and 12 h darkness each day. Every mouse was fed a solid diet (MF, Oriental Yeast Co., Ltd., Tokyo, Japan) and 8–9 week old mice were used in the experiment. Animal experiments were conducted in accordance with the guidelines of the “Guide for the Care and Use of the Laboratory Animals” of Tokyo University of Agriculture and Technology and were approved by the Institutional Animal Care and Use Committee of Tokyo University of Agriculture and Technology (IACUC No. 30-128 and No. R02-130).
2.3. Preparation of a Nanog Overexpressing Cell Line and a TGF-β1 Knockdown Cell Line
An overexpression vector for Nanog was constructed by inserting the Nanog gene into a pCAG-IRES-PuroR-EGFP. The vector product (4 μg/250 μL RPMI) and a Lipofectamine 2000 solution (5 μL/250 μL RPMI) were mixed and incubated at 25 °C for 20 min. The mixture was added to 90% confluent F10 cells and incubated for 3 h. The cells were then cultured in fresh R10 at 37 °C for 48 h. After replacing the medium with R10 containing 1.5 μg/mL puromycin, cells were cultured for 2 weeks to select the Nanog overexpressing cells (Nanog+F10). A knockdown vector for TGF-β1 was prepared by inserting the shTGF-β1 into pU6-PGK-PuroR. Using this vector, a TGF-β1 knockdown cell line was prepared in the same protocol as that for a Nanog overexpressing cell line. The overexpression of Nanog was confirmed by quantitative RT-PCR and western analysis as described below. The knockdown of TGF-β1 was confirmed by quantitative RT-PCR, western analysis, and ELISA as described below.
2.4. Quantitative RT-PCR
Total RNA was prepared by ISOGEN II (Nippongene, Tokyo, Japan) according to the manufacturer’s instructions. The expression levels of
Nanog, EGFP,
puroR,
TGF-β1,
F4/80,
CD68,
CD80,
CD86,
CD163,
and CD206 mRNAs were determined by quantitative RT-PCR using StepOnePlus
TM Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). The analysis was performed under the following conditions: 95 °C for 10 min, 45 cycles of a reaction set (95 °C denaturation for 15 s, 60 °C annealing for 1.0 min), and a reaction set for melt curve analysis (95 °C for 15 s and 60 °C for 1 min). Primer sets of respective target RNAs are listed in
Table S1. The amount of target mRNA was normalized to the amount of
Gapdh mRNA.
2.5. Western Analysis
A protein sample of cells was prepared according to the following procedure. After rinsing 70–80% confluent cells with PBS, an RIPA buffer (25 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, pH 7.6; Thermo Fisher Scientific) was added to the culture dish. The dish was stood on ice for 15 min. Then, the cells were peeled from the culture dish with a cell scraper and collected in a 1.5 mL microtube. The cell suspension was sonicated (UR-20P; TOMY SEIKO, Tokyo, Japan) on ice and centrifugated at 20,000× g for 15 min. The supernatant was collected as a protein sample of cells. A protein solution of EVs was prepared as follows. The pellet of EVs obtained as the precipitate of ultracentrifugation was suspended in an RIPA buffer and stood on ice for 15 min. The protein concentration was determined using a Pierce® BCA ™ Protein Assay kit (Thermo Fisher Scientific). A protein solution was mixed with a 1/6 volume of 0.375 M Tris-HCl (pH 6.8) buffer solution containing 93 μg/mL DTT, 0.12 g/mL SDS, 0.6 mL/mL glycerol, and 0.6 mL/mL bromophenol blue. Then, the solution was heated at 95 °C for 5 min and applied to SDS-PAGE at 150 V.
Blotting onto a PVDF membrane was conducted at 100 V for 3 h at 4 °C. The PVDF membrane was then immersed in a TBS-T (Tris-buffered saline (25 mM Tris, pH 7.4, 150 mM NaCl) containing 1 (
v/
v) % Tween 20) solution containing 5 (
w/
v) % skim milk at 25 °C for 30 min. Then, the PVDF membrane was incubated in a 5% skim milk TBS-T solution containing primary antibody at 25 °C for 3 h. Primary antibodies against mouse Gapdh (1:1000, sc-32233; Santa Cruz Biotechnology, Dallas, TX, USA), mouse Nanog (1:500, ab80892, Abcam, Cambridge, UK), and mouse HSC70 (1:500, sc-7298, Santa Cruz) were used, respectively. After washing with TBS-T three times, the membrane was incubated in TBS-T containing a secondary antibody (anti-mouse immunoglobulin conjugated to alkaline phosphatase, Promega, Madison, WI, USA) at 25 °C for 1 h. Membranes were washed 3 times with TBS-T and then incubated with Western Blue Stabilized Substrate (Promega) for alkaline phosphatase at 25 °C for 5 min. Colored bands of target proteins were quantified using ImageJ software (NIH:
https://imagej.nih.gov/ij/ (Access on 30 October 2017).
2.6. ELISA
The amount of TGF-β1 in EVs and in culture medium were measured using TGF-beta 1 Quantikine ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. After collecting the culture medium from the culture dish, the cells were collected by trypsin/EDTA treatment to estimate the number of cells. The amount of TGF-β1 in the culture medium was divided by the number of cells to determine the amount of TGF-β1 released per cell.
2.7. Measurement of Cell Proliferation
F10 and Nanog+F10 cells were cultured in R10 medium, respectively, in dishes with 6 cm in diameter. The number of starting cells was 1 × 105 cells per dish with 6 cm in diameter. Three dishes were used for the cell count. In total, nine dishes were used for the cell count at 0 h, 48 h, and 96 h. For the dishes for 96 h culture, the medium was replaced at 48 h by fresh R10 for the culture for another 48 h. At respective time points, the cells were harvested by washing with 1 mL PBS twice, treatment with 200 μL trypsin/EDTA for 5 min, adding 2 mL culture medium, and centrifuged at 1500 rpm × 5 min. The precipitate was suspended in 2 mL culture medium and cell count was performed by 8-fold dilutions and with a hemocytometer.
2.8. Wound Healing Assay
Test cells were cultured on a 12-well plate until they became confluent. The monolayer cell sheet on the bottom plate in a well was scratched with a sterile plastic chip to form a model of wound. During culture, the microscopic image of this wound area was recorded from 0 h up to 24 h, and the changes of wound area were analyzed using ImageJ software to determine the wound healing rate.
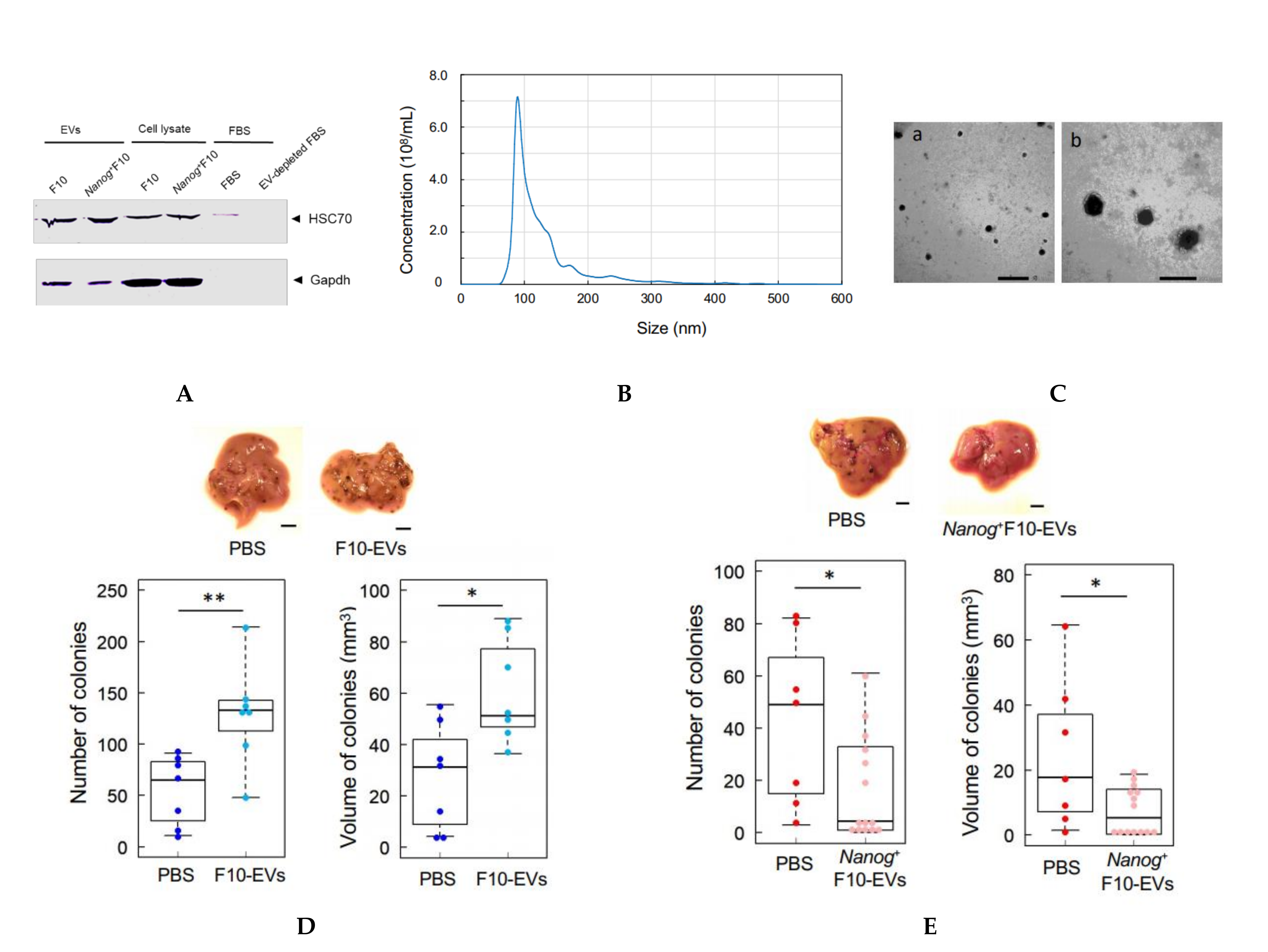
2.9. Isolation of EVs
In this study, HSC70 was used as a marker of melanoma-derived exosomes since it was used in common in both references [
25,
26]. HSC70 was also used as a checking marker of exosome contamination in fetal bovine serum (FBS). FBS was centrifuged at 100,000×
g for 80 min twice to prepare EV-depleted FBS. A trace of contamination of EVs, if any, might interfere with the EVs collected as the HSC70 positive fraction.
The protocol for the separation of EVs was determined by referring to [
27]. Cells were cultured for 48 h and rinsed with PBS. Then, RPMI 1640 medium containing 10% EV-depleted FBS and 1% penicillin streptomycin was added to the culture dish. After the culture for 48 h, the culture medium was collected by centrifugation at 2000×
g for 20 min. The supernatant was centrifugated at 10,000×
g for 40 min and then at 100,000×
g for 80 min. The resulting precipitate was suspended in PBS and passed through a filter with a pore size of 0.22 μm. The filtrate was centrifugated again at 100,000×
g for 80 min, and the final precipitate was suspended in PBS to obtain a suspension of EVs.
2.10. Analysis of Size Distribution of EVs
The size distribution of EVs was analyzed using a NanoSight NS300 system (Malvern Panalytical, WR14 1XZ, Malvern, UK). A 100 μL PBS containing more than 107 particles was prepared and analyzed. The mode diameter and mean diameter were determined by five measurements.
2.11. Observation of EVs with an Electron Microscope
The same amount of 4% paraformaldehyde (PFA) and a PBS suspension of EVs were mixed and incubated at 25 °C for 30 min for fixation of EVs. Three μL aliquots of the fixed suspension of EVs were placed dropwise on a grid with a support membrane (JEOL, Tokyo, Japan) that was made hydrophilic and dried at room temperature in advance. The grid was washed with PBS 7 times, fixed with 1% glutaraldehyde for 5 min, washed with pure water 7 times, stained with a 3% phosphotungstate solution for 10 min, and dried at room temperature for 10 min. The sample of EVs on the grid was observed with a transmission electron microscope.
2.12. Count of Metastatic Colonies and Estimation of Volume
Melanoma cells (2.5 × 105 cells/250 μL PBS) were injected into the tail vein of 8–9 week old C57BL/6 male mice. Two weeks later, mice were euthanized by cervical dislocation, and the livers were separated into lobes and photographed under a microscope. EVs (5 μg/100 μL PBS) were injected into the tail vein of 5–6 week old mice three times per week for 3 weeks, and subsequently melanoma cells (2.5 × 105 cells/250 μL PBS) were injected into the tail vein. Ellipsoidal major and minor diameters of each metastatic colony were analyzed using ImageJ software, and the volume was calculated by using the following formula; V = 1/6πab2, where, a and b are the major and minor diameters, respectively.
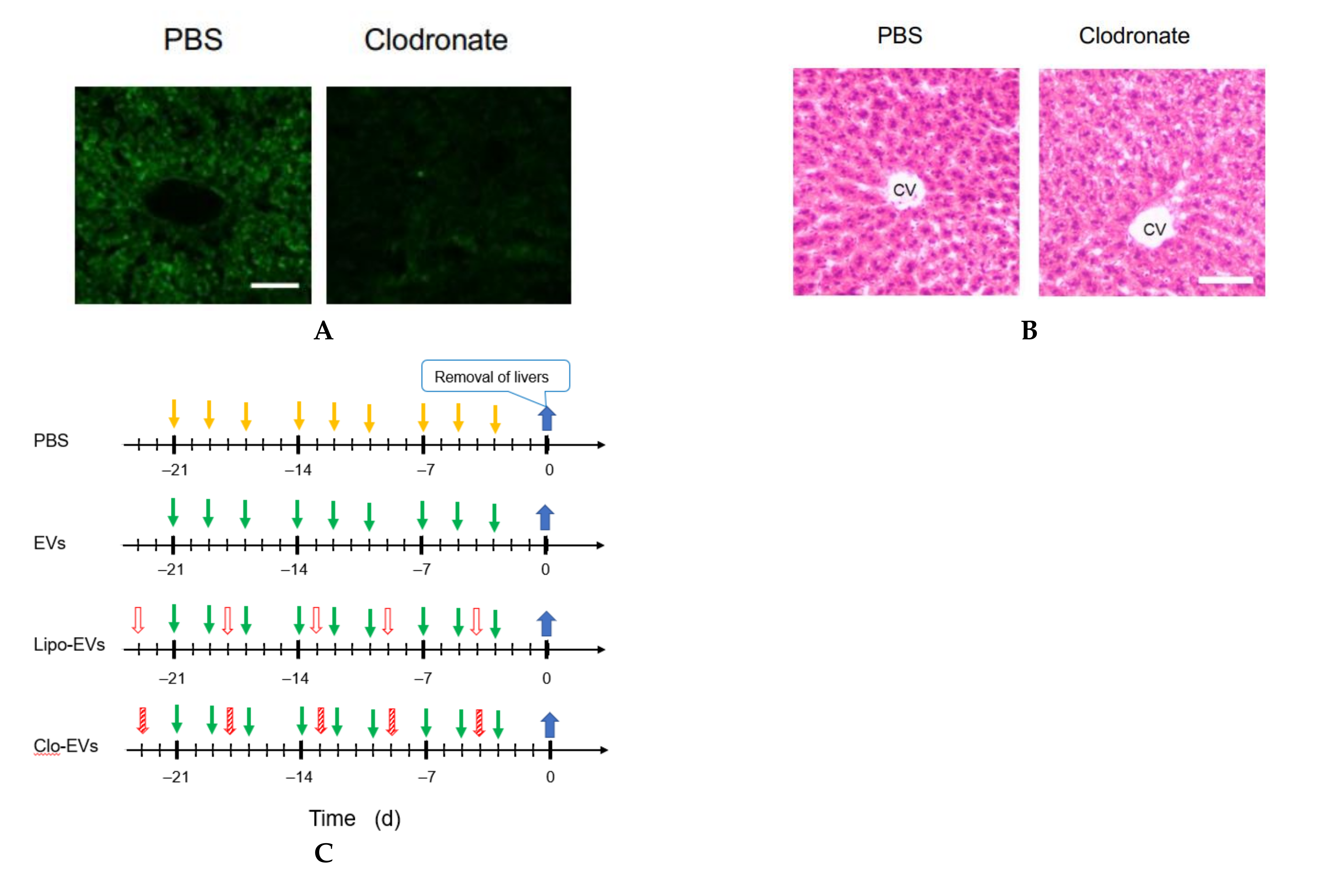
2.13. Macrophage Depletion
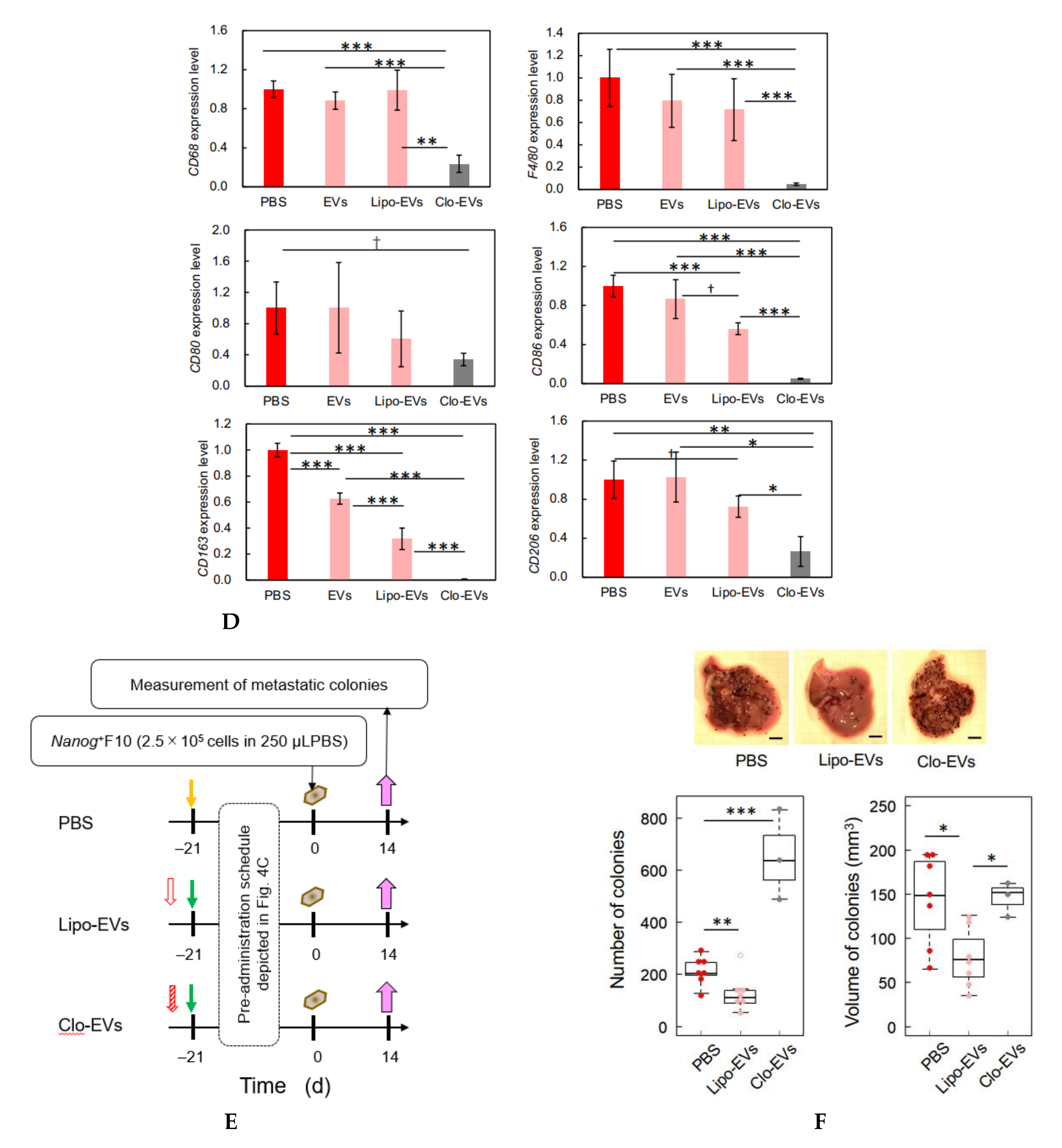
Clodronate was used to deplete the function of macrophages. Clodronate-encapsulated liposomes (12.5 mg/kg mouse weight) (Hygieia Bioscience, Osaka, Japan) were repeatedly injected into the tail vein of mice for 3 weeks at an interval of 4 or 5 d. The effect of clodronate was confirmed by the decrease or loss of the gene expression of 6 macrophage markers and histochemical analysis by hematoxylin-eosin (HE) staining.
2.14. HE Staining
A liver of the mouse was embedded in Neg-50 (Thermo Fisher Scientific) and allowed to stand at 4 °C for 18 h. Frozen blocks were prepared using isopentane (FUJIFILM Wako Pure Chemical, Osaka, Japan) immersed in liquid nitrogen. The frozen block was sliced into 7 μm thick tissue sections using a cryostat HM550OVP (Thermo Scientific Microm) and a MAS-coated slide glass (MATSUNAMI, Kishiwada, Osaka, Japan) was pressed against the tissue section for its attachment on the glass. The frozen tissue section on the glass was washed with water and immersed in Meyer-hematoxylin solution (FUJIFILM Wako Pure Chemical) for 10 min for nuclear staining. Subsequently, it was washed with running water for 5 min and immersed in an eosin solution for 2 min to stain the cytoplasm. It was rinsed with water for about 5 s to wash off excess eosin solution, and soaked in 70 (v/v) % ethanol for 5 min, then in each of 90, 95, and 100 (v/v) % ethanol for 2 min in this order. After that, it was soaked in xylene for 5 min twice, and encapsulated with a specimen-encapsulating agent New MX (MATSUNAMI), a xylene-based anti-fading agent.
2.15. Fluorescent Immunostaining of CD68
Frozen tissue sections were fixed by dropping 4% PFA and allowing to stand for 15 min. It was then washed by immersing it in 10 mM glycine/PBS three times for 5 min. A 2% gelatin solution was added dropwise to the tissue section and allowed to stand for 20 min for blocking. The washing was repeated 3 times by immersing in 10 mM glycine/PBS for 5 min. After further immersing in 0.1% BSA/PBS for 5 min, 1% BSA/PBS containing an antibody against CD68 (1:50, sc-20060, Santa Cruz) was added dropwise, and the mixture was allowed to stand for 40 min. The washing was repeated 5 times by immersing in 0.1% BSA/PBS for 5 min. Next, 1% BSA/PBS containing goat anti-mouse IgG H&L labelled with Alexa Fluor 488 (Ex: 496 nm, Em: 519 nm) (1:200, ab150113, Abcam) was added dropwise and allowed to stand for 40 min. The washing was repeated 5 times by immersing in 0.1% BSA/PBS for 5 min. Counterstaining was performed by immersing in a hematoxylin solution for 30 s and rinsing with a running water for 5 min. Moisture around the section was wiped off with a Kimwipe, dried for about 1 min, then the encapsulant New MX was added dropwise, and the mixture was sealed with a cover glass. The sample was observed with an inverted confocal microscope.
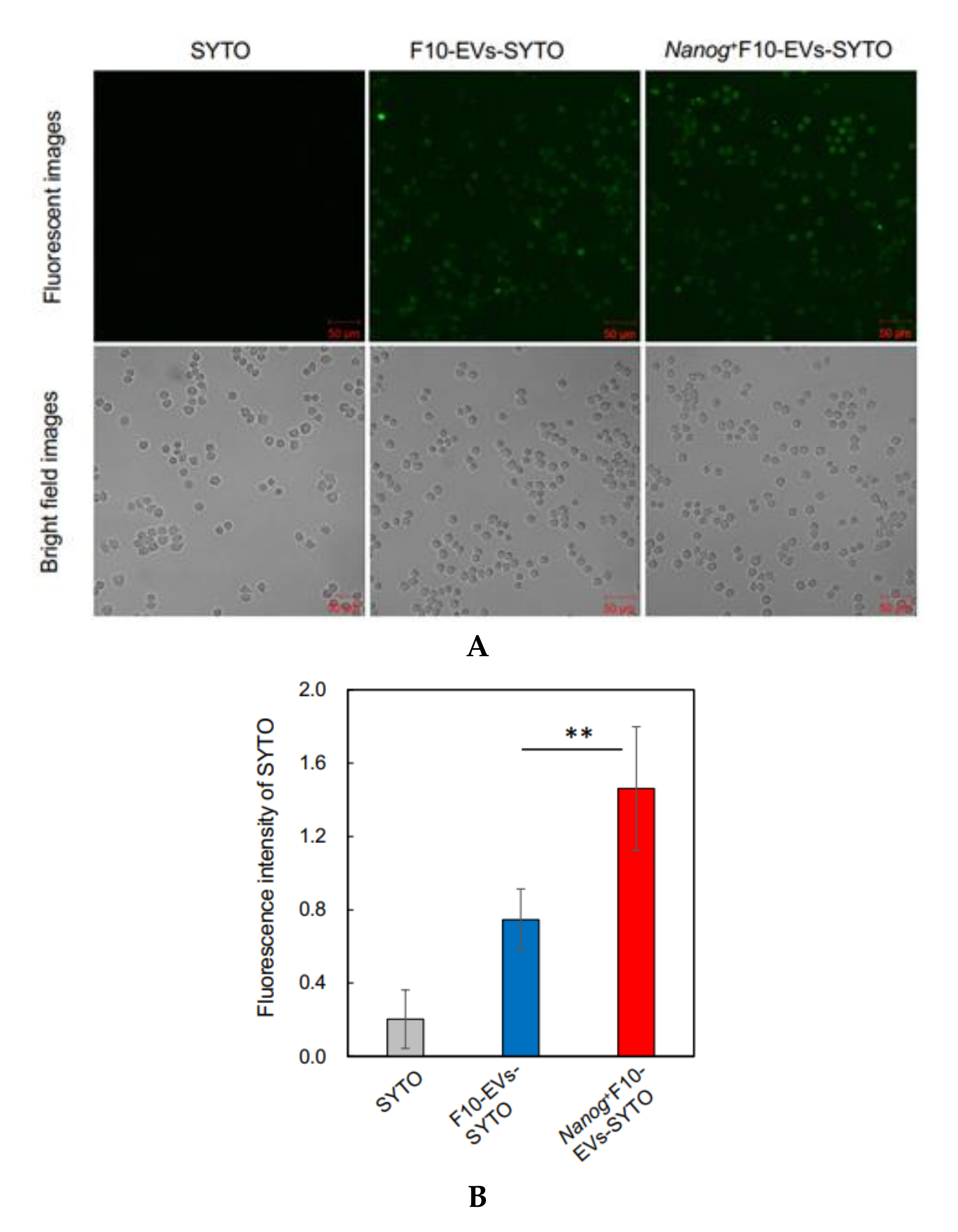
2.16. Fluorescence Analysis of the Uptake of EVs by J774.1 Cells
EVs were labelled with SYTO RNASelect™ Green Fluorescent cell Stain (SYTO) (Thermo Fisher Scientific) (Ex: 490 nm, Em: 530 nm) by incubation at 37 °C for 20 min in the dark. A mouse macrophage cell line, J774.1 cells were seeded in a 6-well plate at 2 × 105 cells/well in 2 mL of R10 medium. The next day, cells were washed twice with PBS and 2 mL of EVs-depleted medium was added to the well. Then, 60 μg of fluorescently labeled EVs (F10-EVs-SYTO or Nanog+F10-EVs-SYTO), SYTO (EVs-less control), or PBS (no treatment control) were added to the well. After incubation at 37 °C for 1 h in the dark, the cells were washed twice with PBS and covered with 2 mL of EVs-depleted medium for the fluorescence image analysis with a confocal microscope. A rectangular area was arbitrarily selected from a microscopic image. The total area of cells (Scell) was determined from the bright field image. The total fluorescent intensity (Fcell) was determined by integrating the fluorescent intensities over Scell. The fluorescent intensity per unit area for this image was determined by Fcell/Scell. Five images were analyzed for each of four conditions (F10-EVs-SYTO, Nanog+F10-EVs-SYTO, SYTO, PBS). The Fcell/Scell value for PBS condition was subtracted from those for the other conditions.
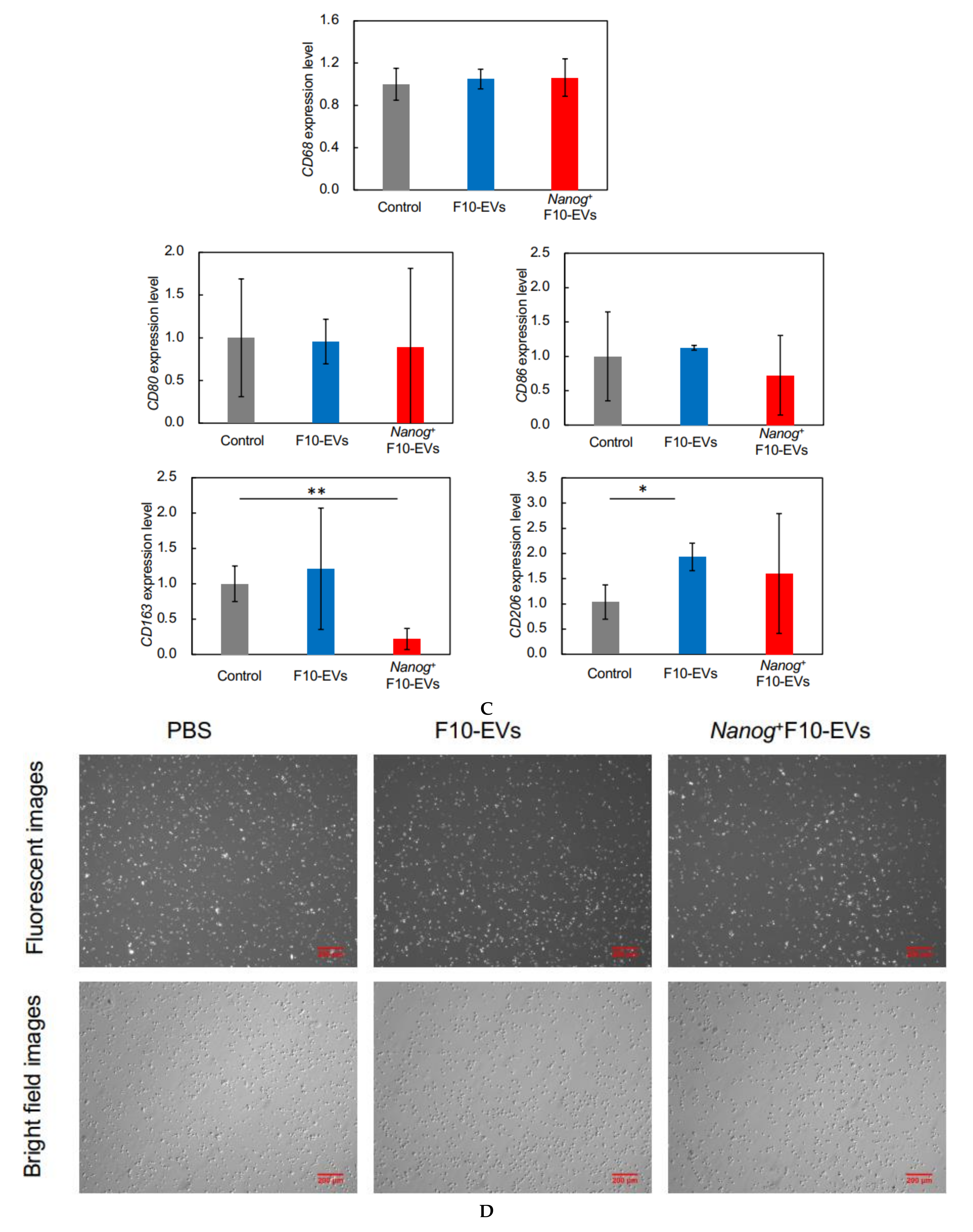
2.17. Fluorescent Immunostaining of CD163 in J774.1 Cells
J774.1 cells were cultured at 2.5 × 105 cells/dish with 20 μg of F10-EVs, Nanog+F10-EVs, or PBS (control) for 48 h. The cells were collected by trypsin/EDTA treatment and suspended in PBS. An aliquot of cell suspension containing 1.0 × 106 cell was collected and washed twice with PBS containing 3% BSA. Then, the cells were reacted with anti-CD163 antibody (1:10, sc-58965, Santa Cruz) at room temperature for 1 h and subsequently with goat anti-mouse IgG (H + L) Alexa Fluor 568 (1:285.7, A-11004, Invitrogen, Waltham, MA, USA) at room temperature for 30 min. The cells were plated on a glass dish and the fluorescent images were analyzed using ImageJ. The fluorescent intensity and area of every fluorescent cell was integrated for an image to determine the fluorescent intensity per unit area. Eight or nine images were analyzed for every condition and the results of F10-EVs and Nanog+F10-EVs were expressed as relative values to that of PBS.
2.18. Statistics
Specific details regarding statistical analyses are presented in the figure legends. Preparation of test samples for mRNA or protein was done using one test sample per dish. Each test sample was analyzed twice or three times and the average of the two or three results was recorded as the value for one test sample. Results are presented as mean ± standard deviation (SD) or mean ± standard error of mean (SEM) for the number of samples (n). Results of metastatic colony analyses are presented in box plots. Outliers shown in box plots were determined by a Smirnoff–Grubbs test to be greater than 0.05 one-tailed probability. The statistical significance between two specific data groups was analyzed by two-tailed Student’s t test. The statistical significance of results is denoted by a p value or by marking with asterisk(s): ***: p < 0.001, **: p < 0.01, *: p < 0.05, †: p < 0.1.
4. Discussion
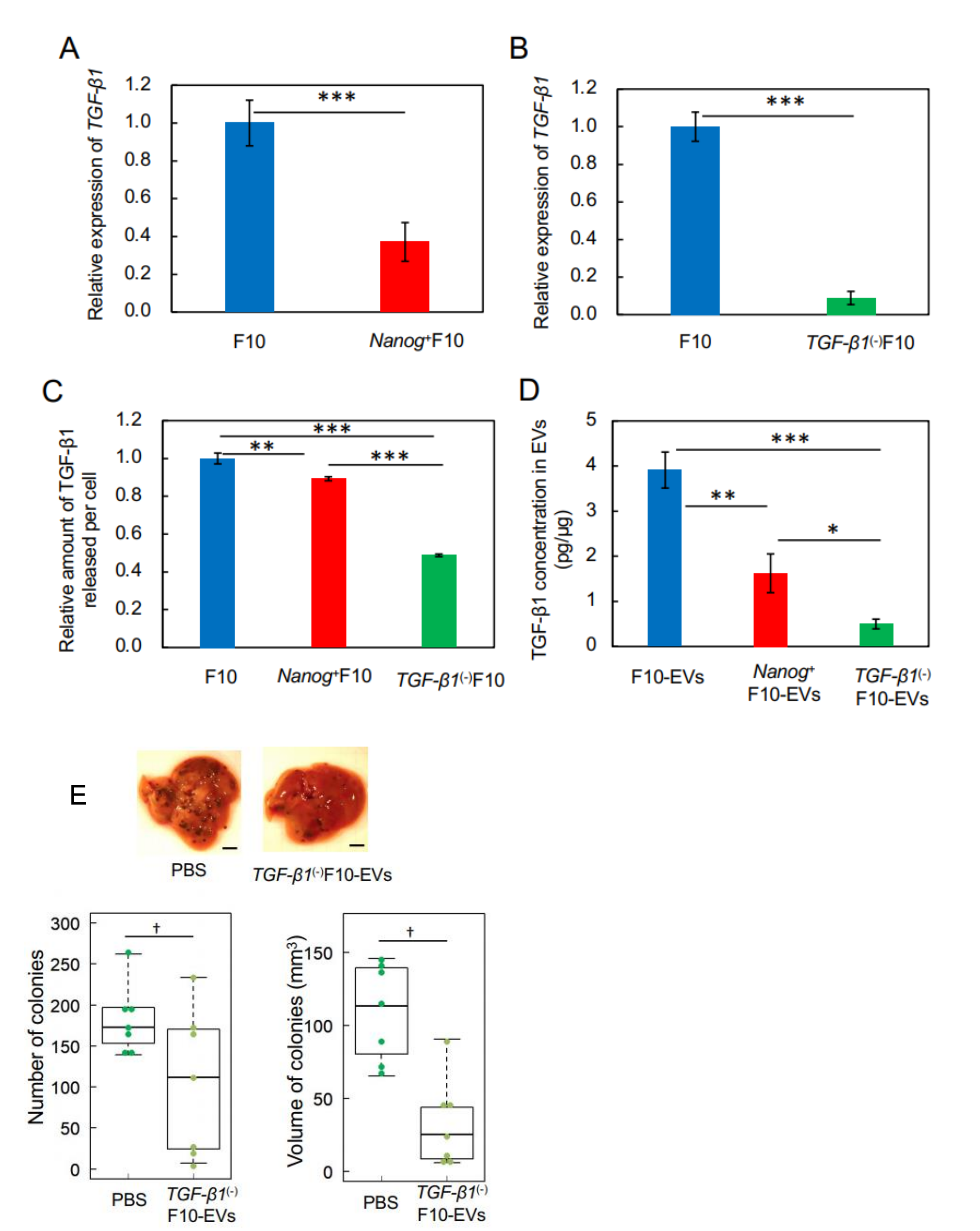
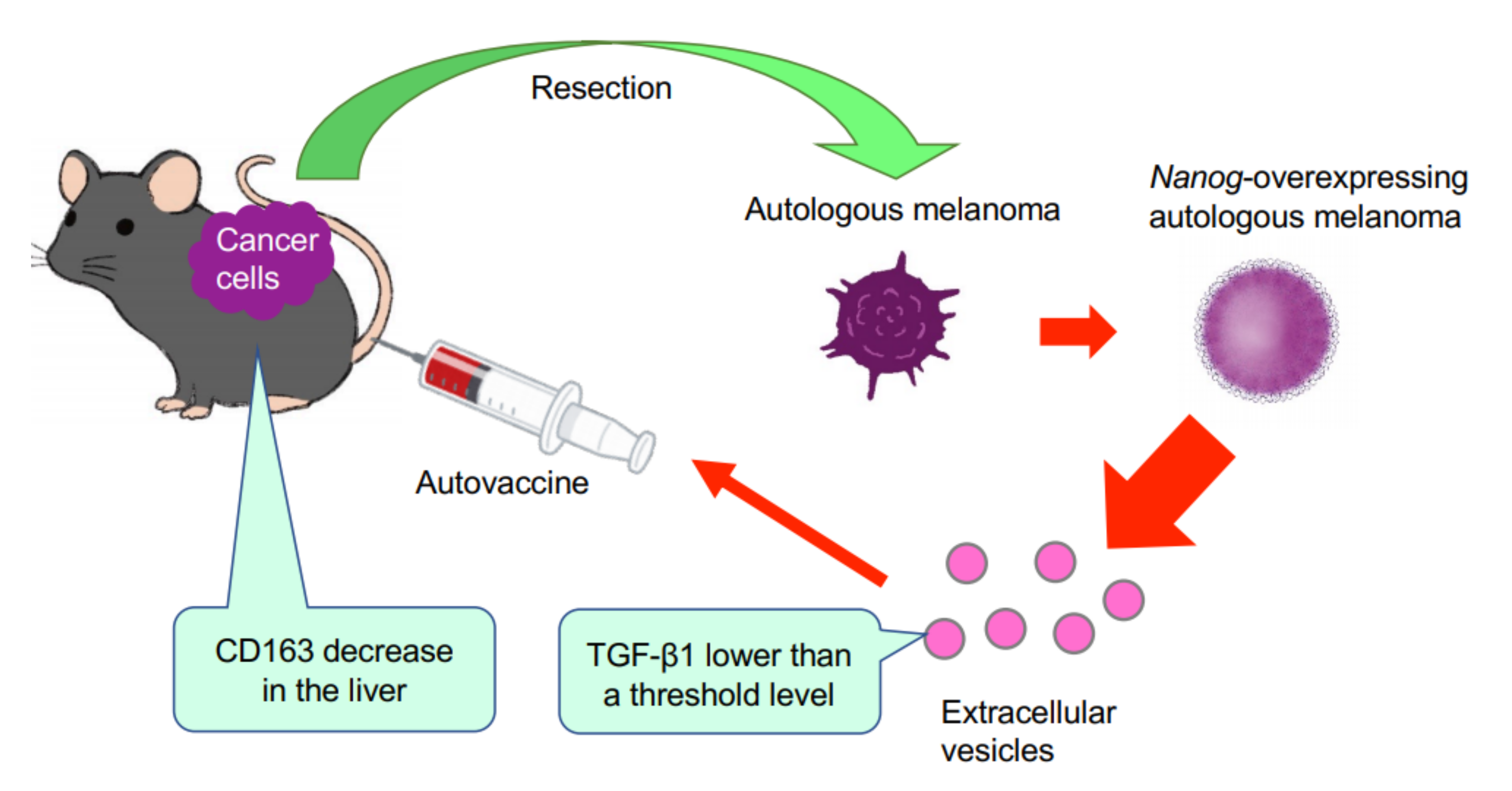
This study has demonstrated for the first time that EVs derived from cancer cells with high metastatic potential is an autovaccine candidate based on the concept depicted in
Figure 6. An important key factor was
Nanog overexpression that down-regulated
TGF-β1. The decrease in TGF-β1 was observed in
Nanog+F10 cells as compared to that in F10 cells. The decrease in TGF-β1 was also observed in EVs derived therefrom. The effects of EVs on the metastasis, however, were exactly opposite for F10-EVs and
Nanog+F10-EVs. The metastasis-suppressive effect obtained with
Nanog+F10-EVs was a surprising finding. Since the concentration-dependent opposite effects were observed, it seemed to be a dual role of TGF-β1.
It has been well discussed that the dual role of TGF-β1 is associated with factors that depend on the conditions of the microenvironment. TGF-β1 is tumor-suppressive in tumors in early stage, but tumor-promotive in advanced cancers. TGF-β1 suppresses ID1 (inhibition of differentiation) in normal cells, but promotes ID1 in cancer cells [
19,
30]. Activated ID1 promotes metastasis via progression of epitherial–mesenchymal transition (EMT). However, it is unclear what factors are crucial in the difference between microenvironments in normal or early stage tumor cells and in advanced cancer cells. The number of cancer cells and the concentration of TGF-β1 generated thereby might be one of those factors of difference. The concentration-dependent role of TGF-β1 is also suggested from the morphogen like function of TGF-β1 because TGF-β1 is a member of a morphogen superfamily TGF-β [
31].
We thought that clearer experimental results should be necessary to claim the concentration-dependent role of TGF-β1. Then, the concentrations of TGF-β1 in F10-EVs and
Nanog+F10-EVs were analyzed and could be determined as 3.9 pg/μg and 1.6 pg/μg, respectively. This suggested the presence of a threshold concentration between 1.6 pg/μg and 3.9 pg/μg; metastasis-suppressive below the threshold and metastasis-promotive above the threshold concentration. Experimental results of TGF-β1 knockdown supported the metastasis-suppressive effect at a concentration lower than the threshold. Although there are few papers that report quantitative studies on the role of TGF-β1 in EVs, we have found a couple of papers that may support the validity of the presence of such a threshold level. Exosomes derived from melanoma A375 cells contained 10–15 pg/μg TGF-β and inactivated T-cells, suggesting a metastasis-promotive role [
22]. In contrast, EVs derived from murine colon carcinoma cells that had been genetically modified with overexpression of shRNA for TGF-β1 could induce the tumor growth inhibition [
16]. This suggests a metastasis-suppressive effect at a sufficiently low level of TGF-β1.
Since TGF-β1 is involved in immunosuppression via the induction of tumor promotive M2-like macrophages [
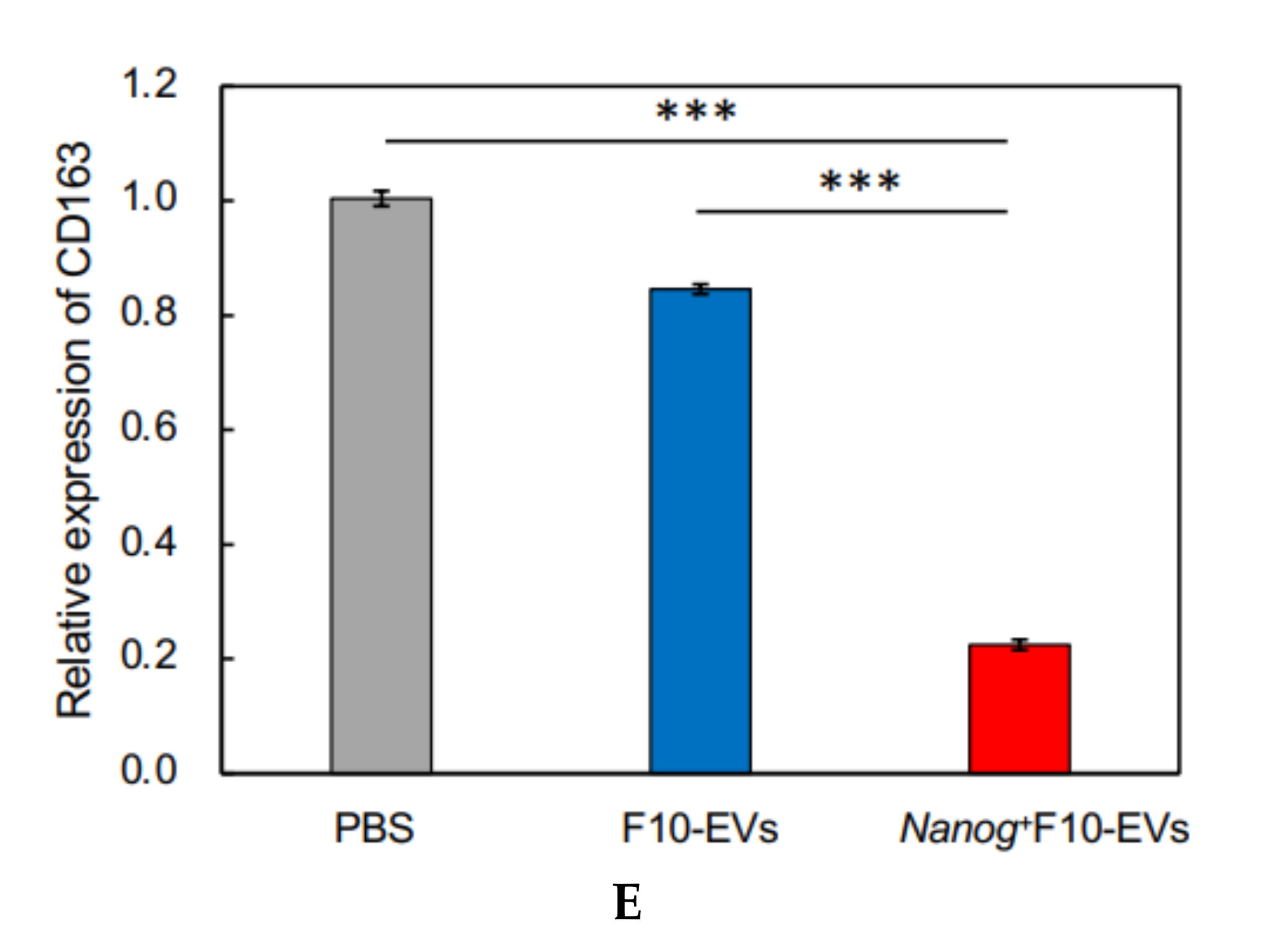
23], we investigated the responses of six markers selected from three categories of macrophages; pan-macrophage, M1-type, and M2-type macrophage markers. Only CD163, an M2-type macrophage marker, showed a significant decrease in gene expression (
Figure 4D) and in protein level (
Figure 5C). CD163 positive macrophages are a subpopulation of M2-type macrophages [
32]. Proliferation of human sarcoma cells is promoted in co-cultures with CD163-positive macrophages, but not with si-
CD163 [
33]. This effect of CD163 is brought about via the production of tumor cell growth factors such as IL6 and CXCL2. IL6 promotes tumor cell proliferation, survival, and metastasis through activation of Stat3 [
34,
35]. Therefore, the decrease of CD163 is suggestive of a metastasis-suppressive response, and such a response is consistent with the
Nanog-induced decrease in TGF-β1 in EVs below the threshold level.
Our next study will be directed towards the further fractionation of Nanog+F10-EVs to purify active components, and the survey of other types of responding immune cells.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








