X or Y Cancer: An Extensive Analysis of Sex Differences in Lung Adenocarcinoma



Abstract
:1. Introduction
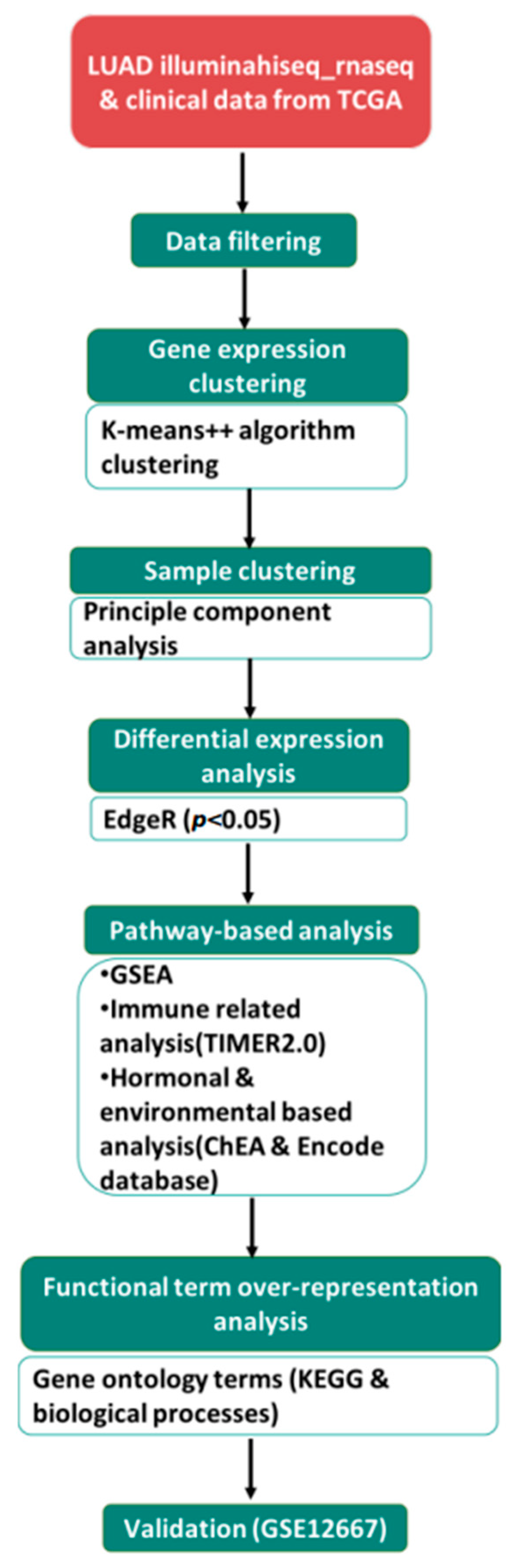
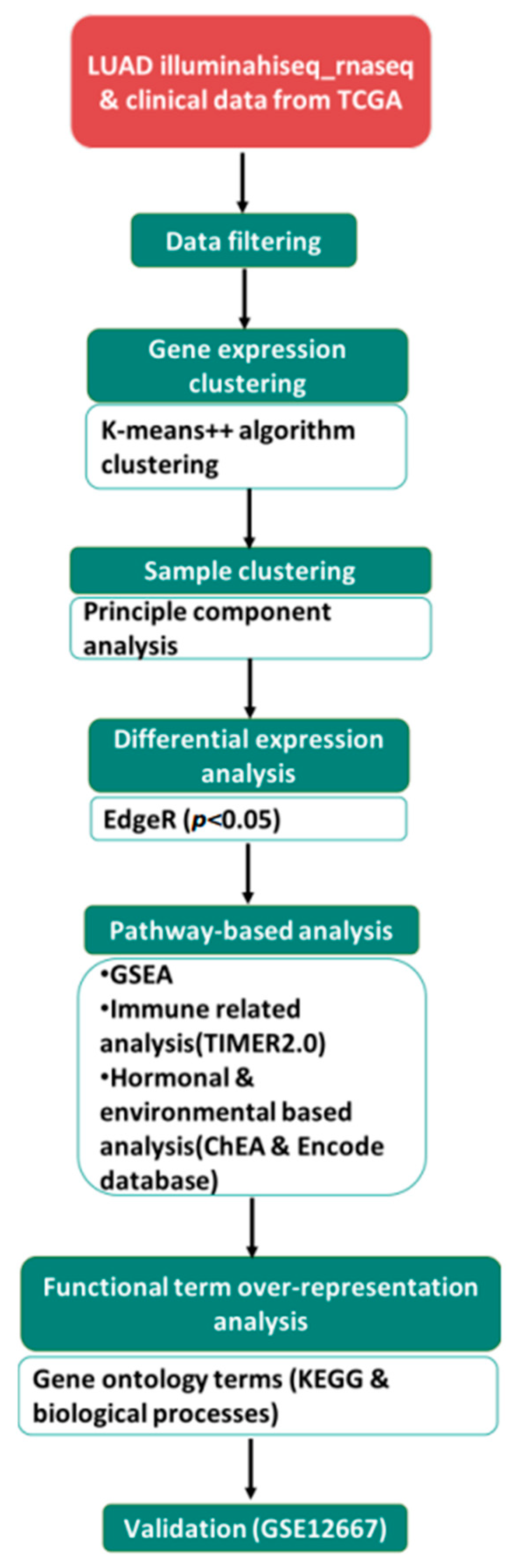
2. Materials and Methods
2.1. Data Collection
2.2. Defining a Gene List of Interest and Identifying Regulatory Networks
2.3. Gene Enrichment Analysis of DEGs
2.4. Correlation of DEGs with Immune Infiltrates
2.5. Statistical Analysis
2.6. Association Analysis between DEGs and Patient Prognosis
3. Results
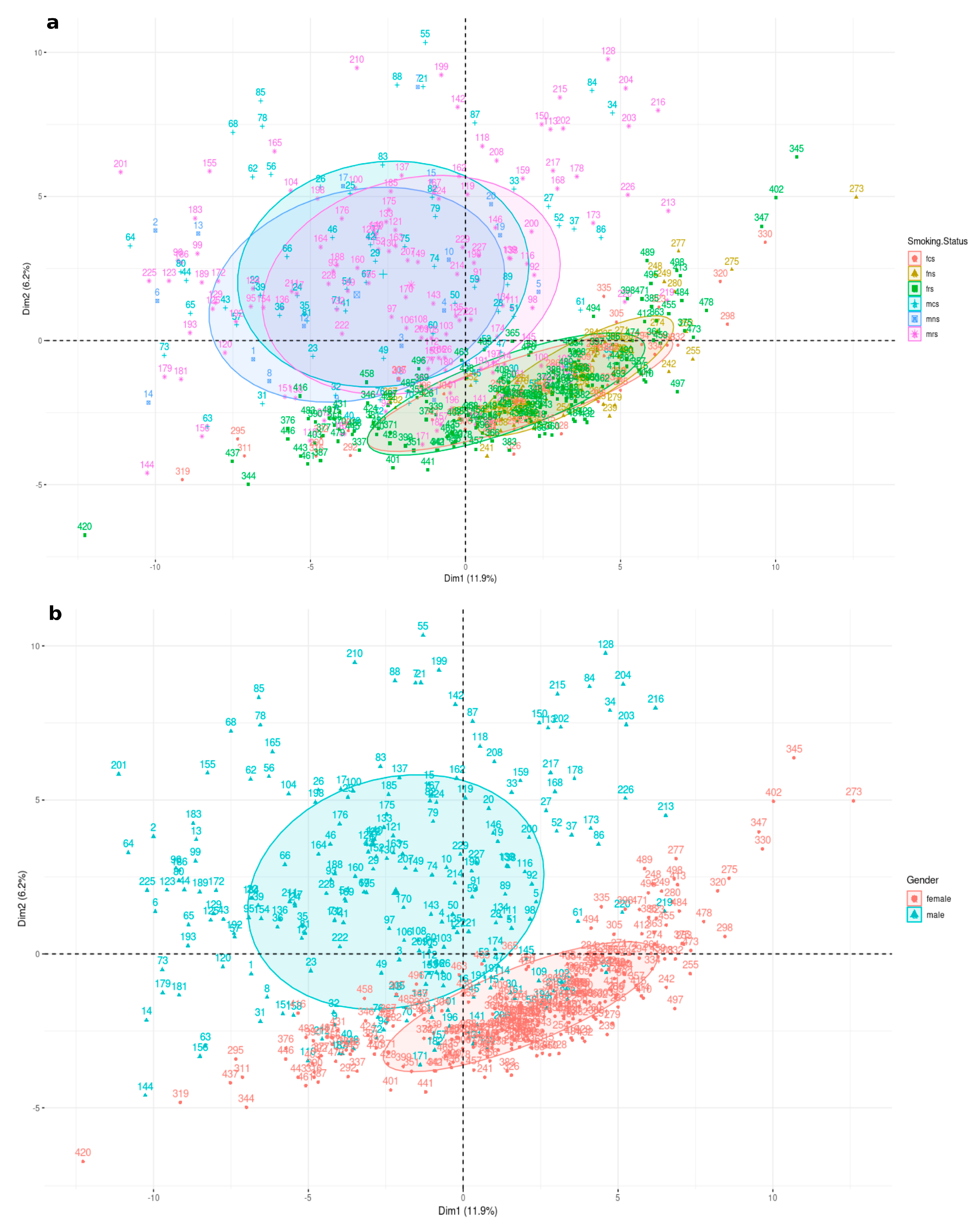
3.1. Genetic Differences Analyses
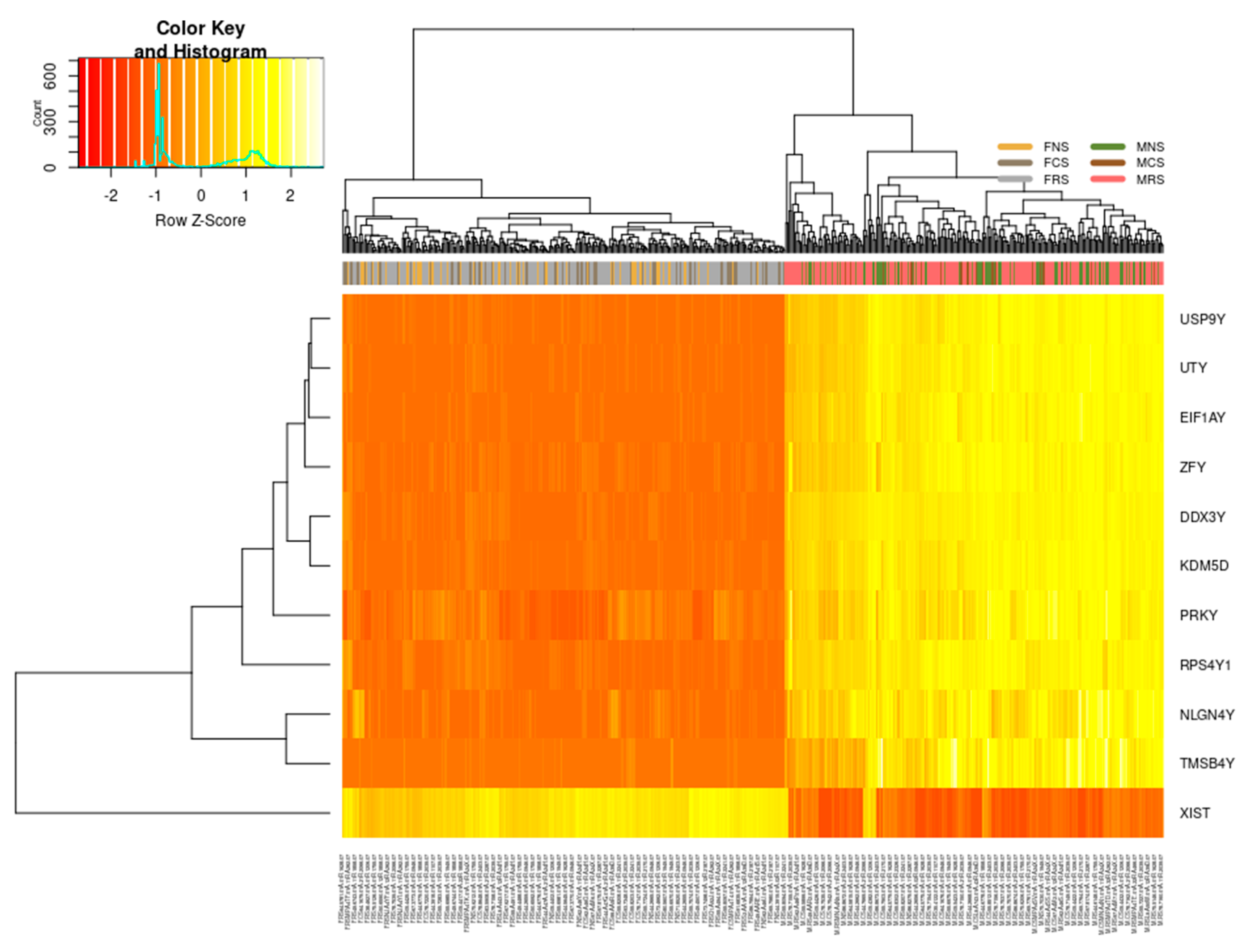
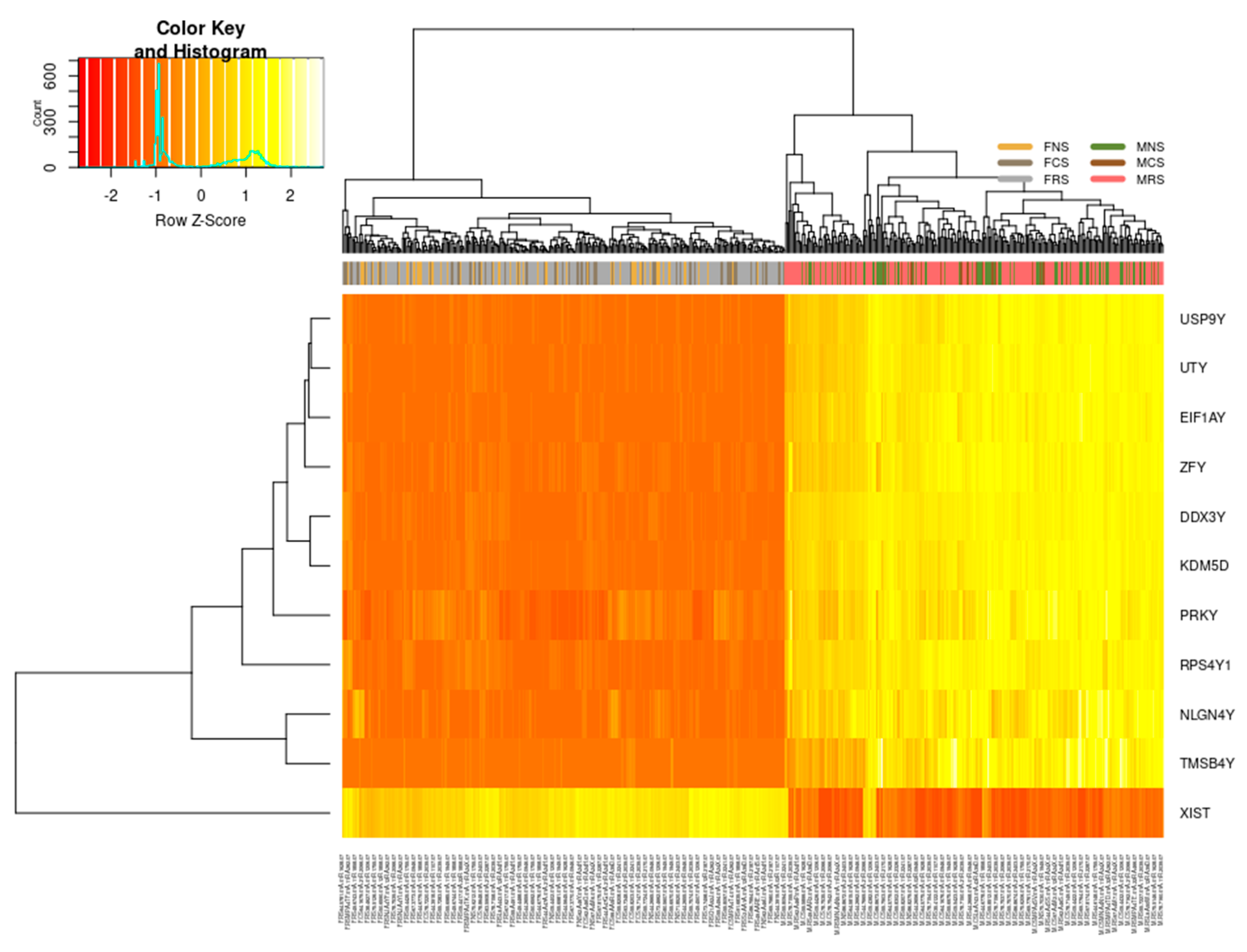
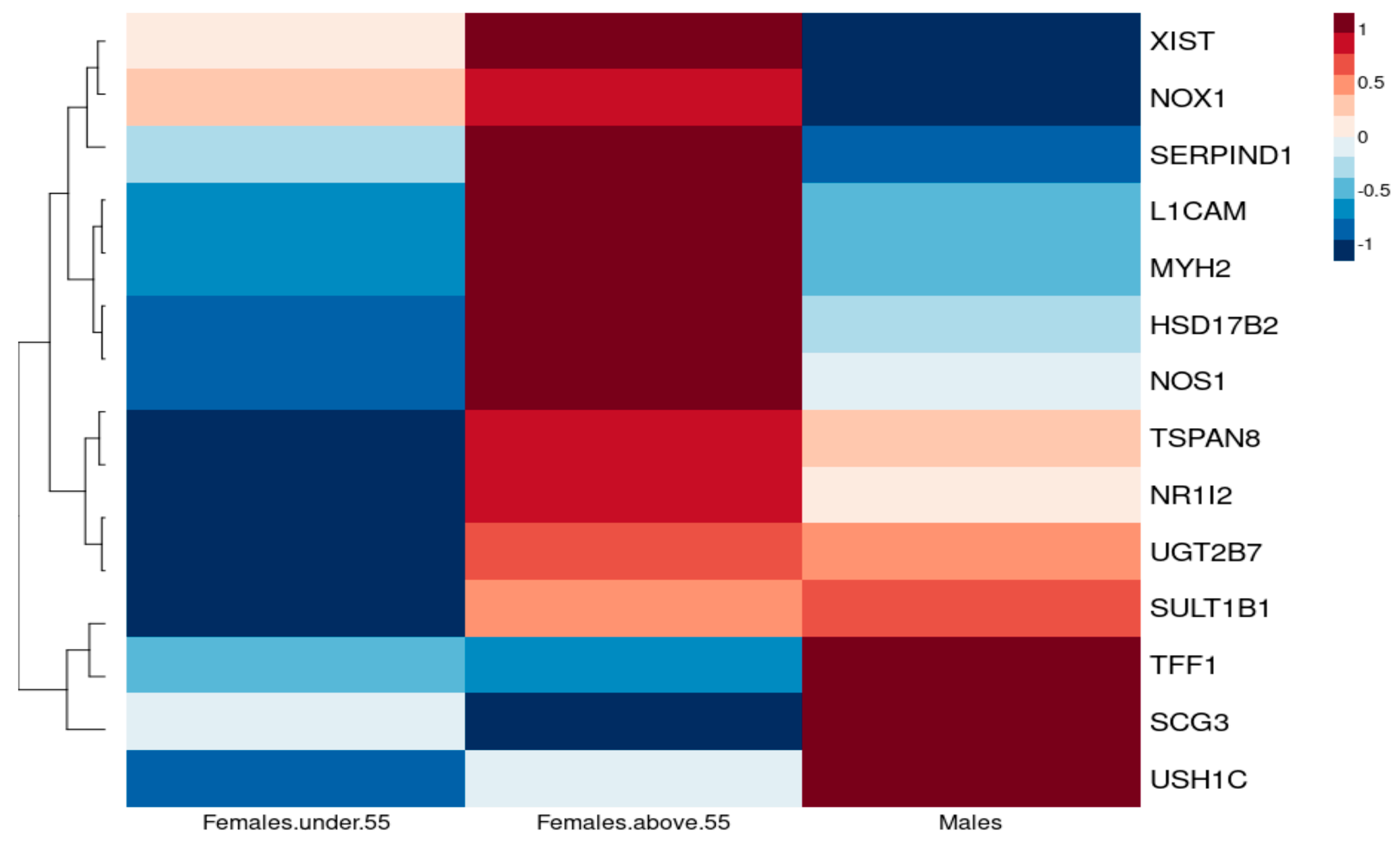
3.1.1. Sex-Specific Molecular Signature of LUAD
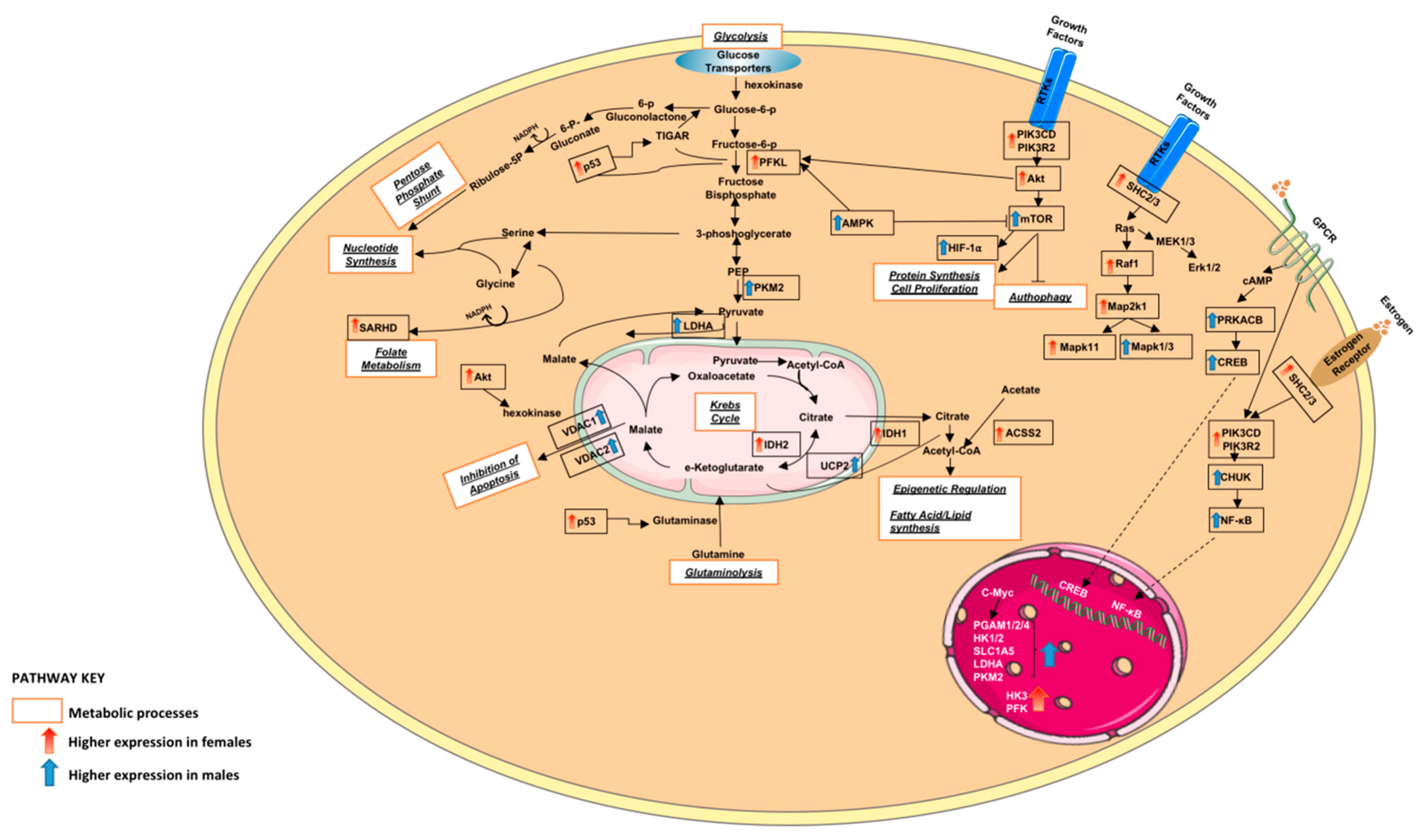
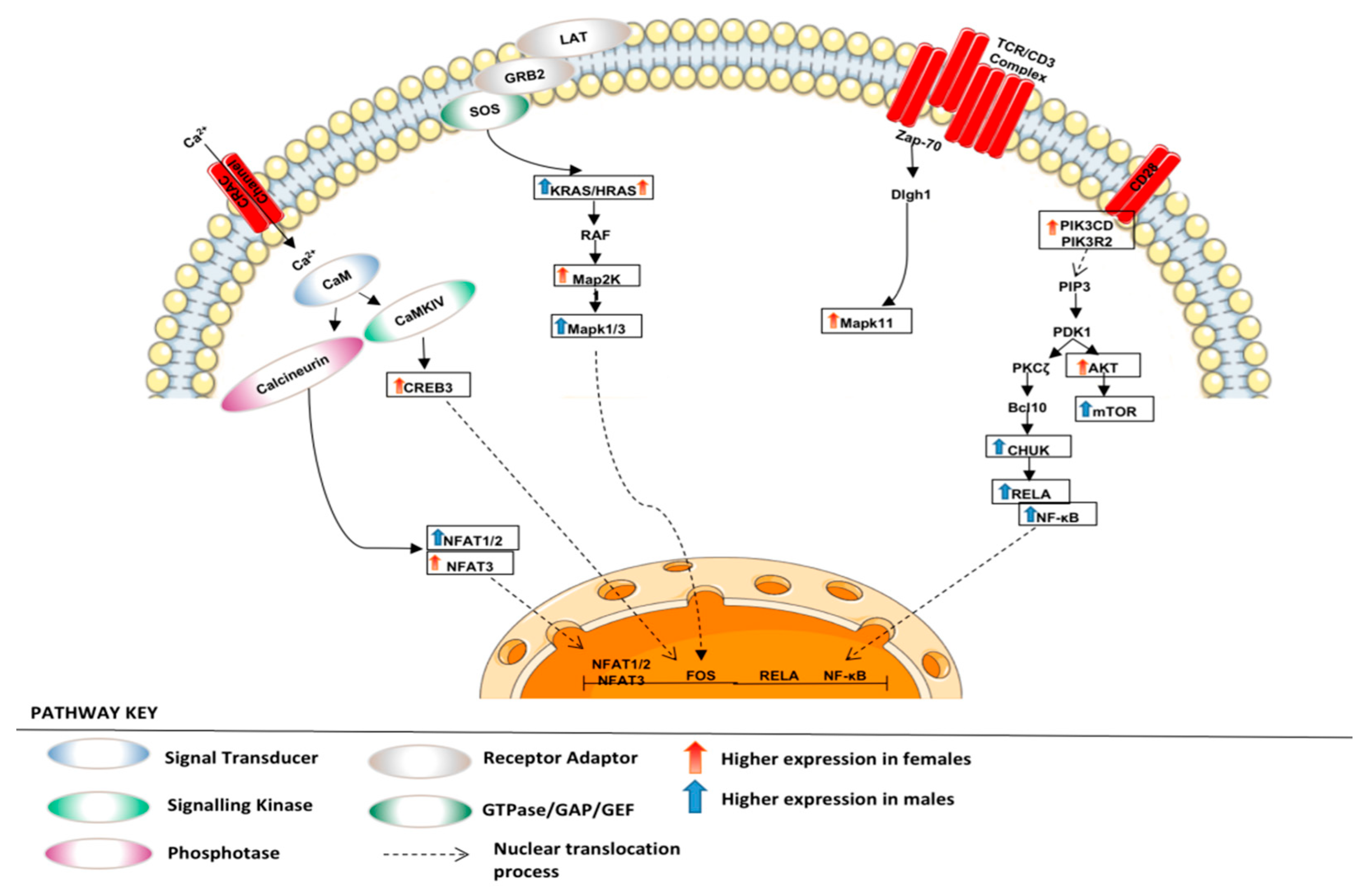
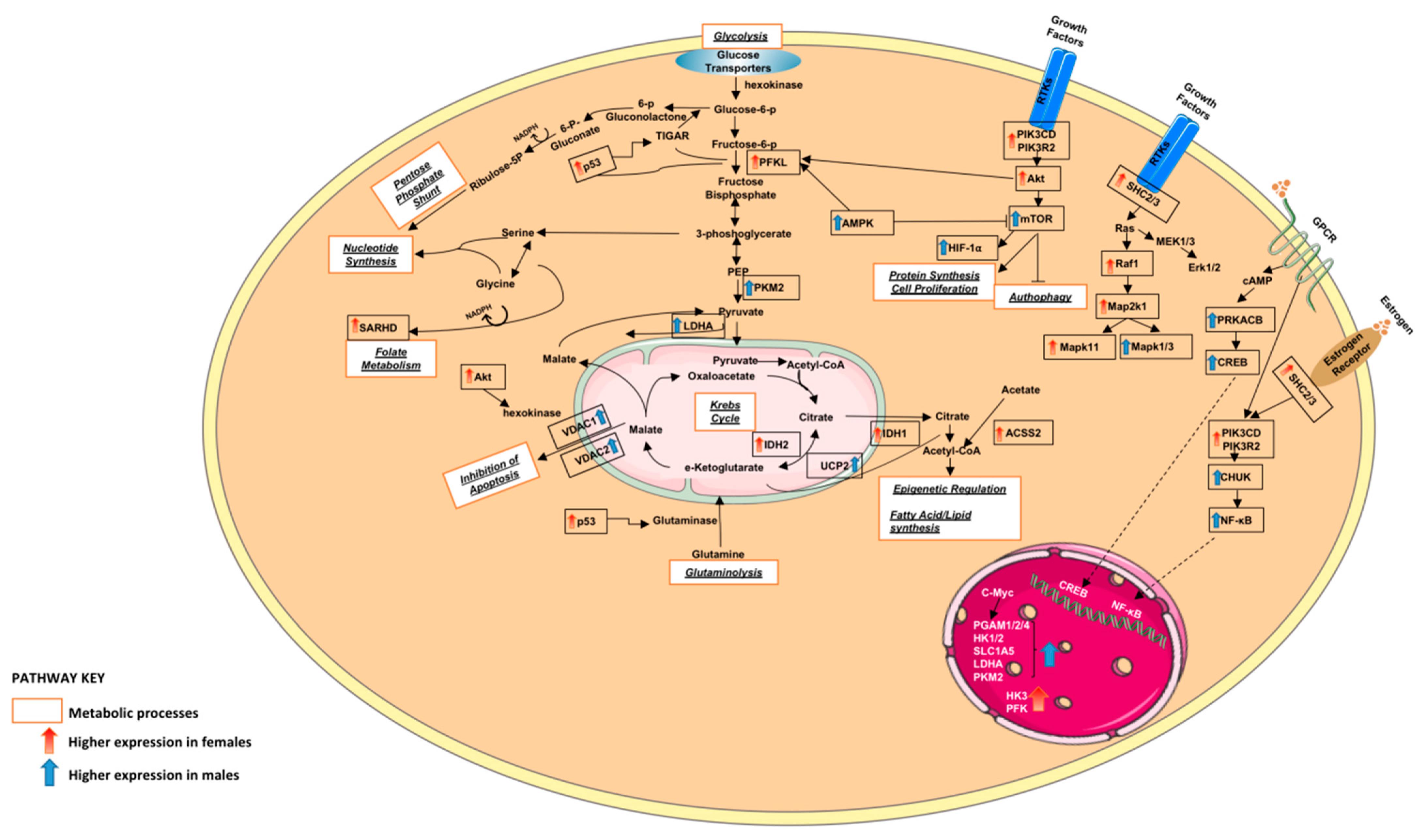
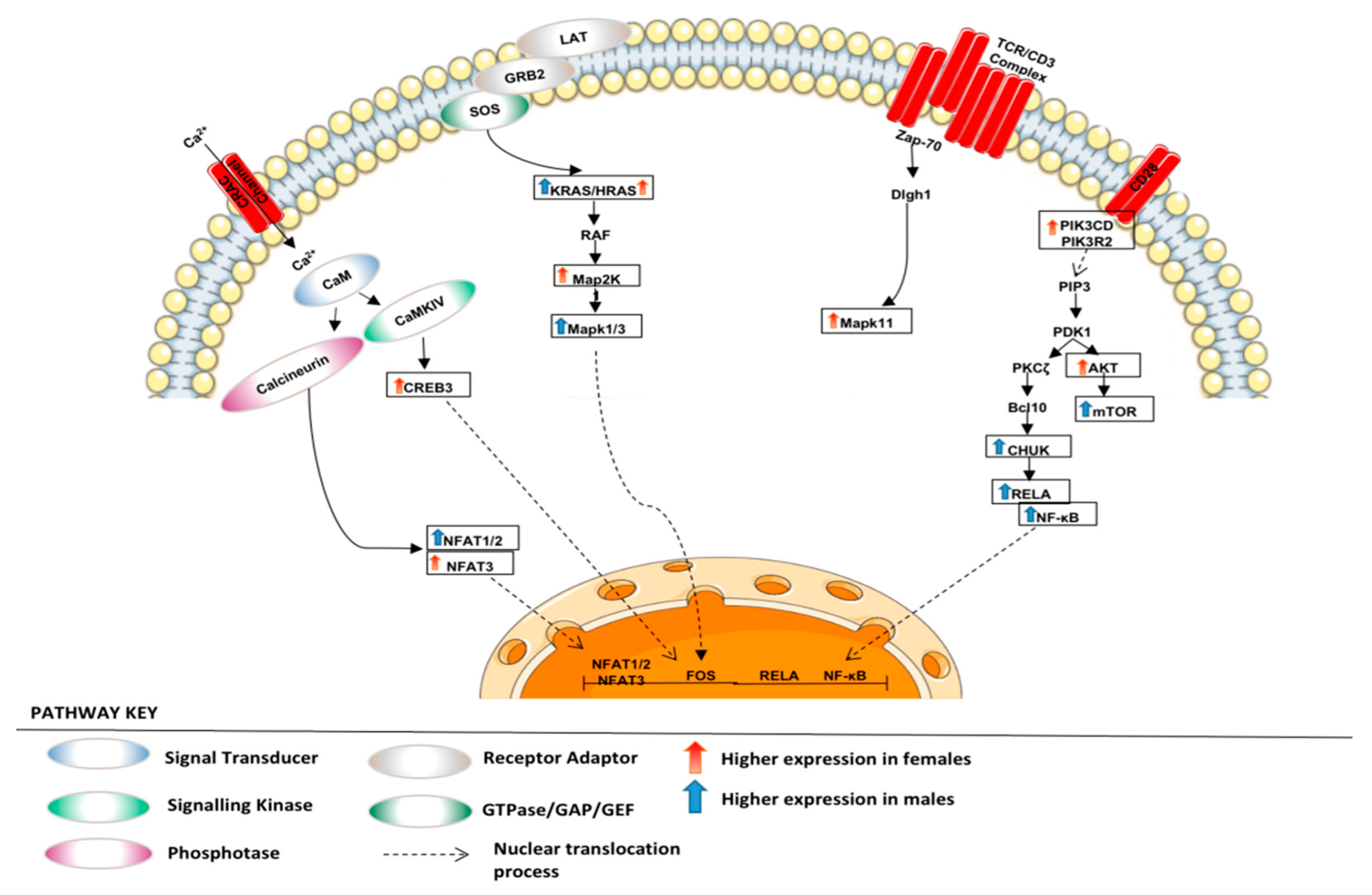
3.1.2. Altered Warburg Metabolism Seems to Inhibit AMPK but Activate mTOR and MAPK Signalling in Our Female Cohort
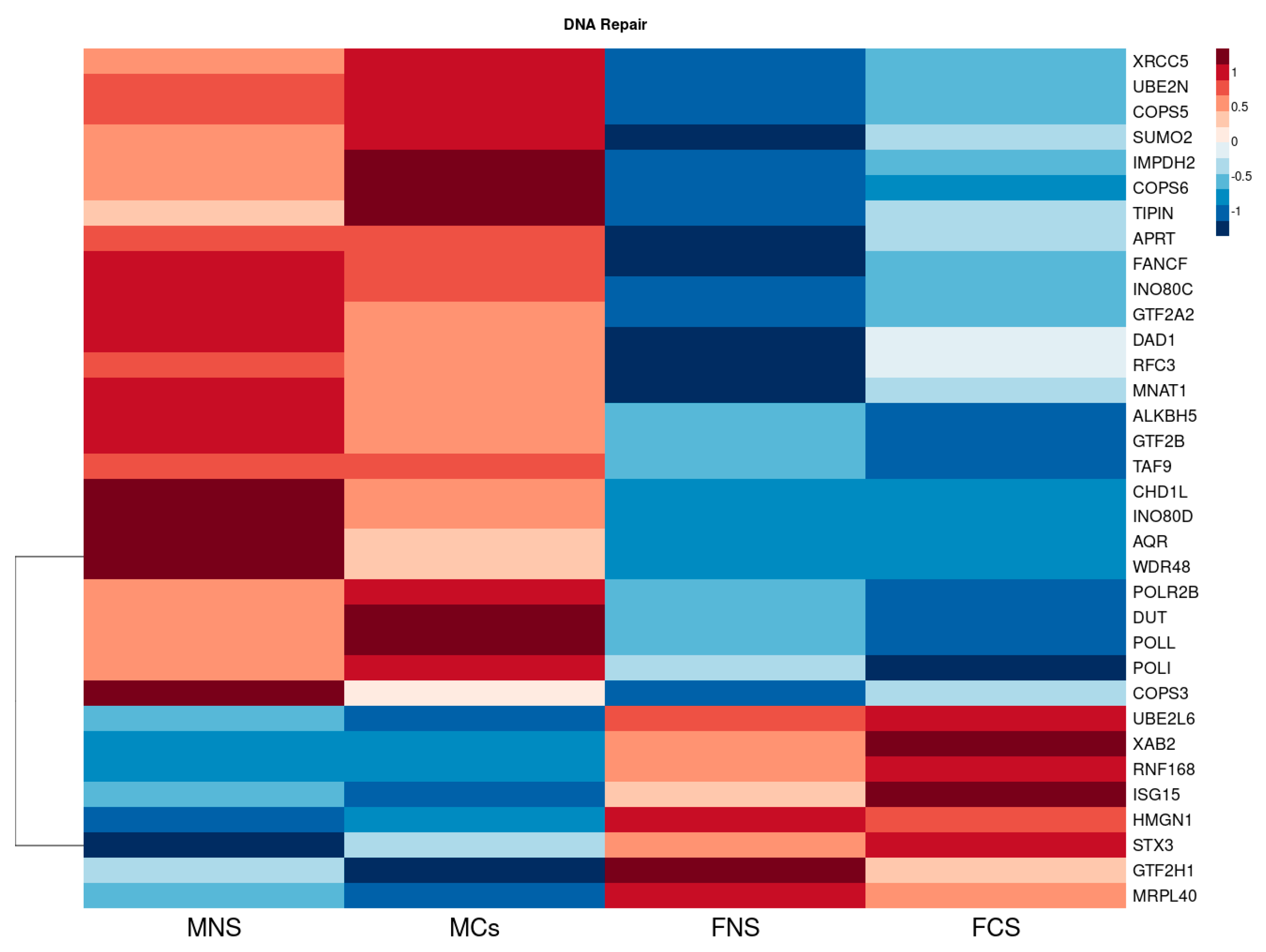
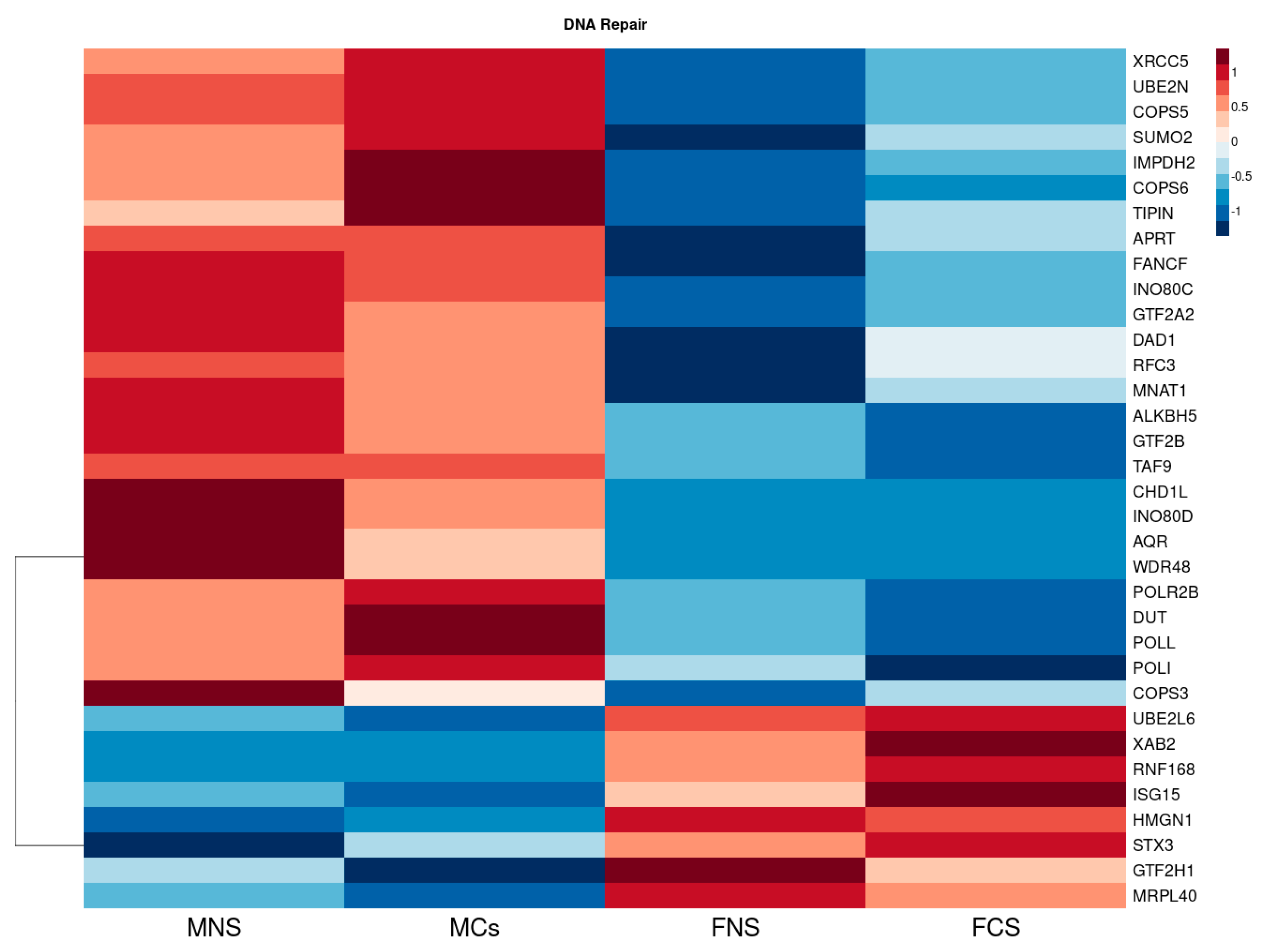
3.1.3. Metabolism of Xenobiotic by Cytochrome p450 and DNA Repair Seem to Be Upregulated in Our Male Cohort
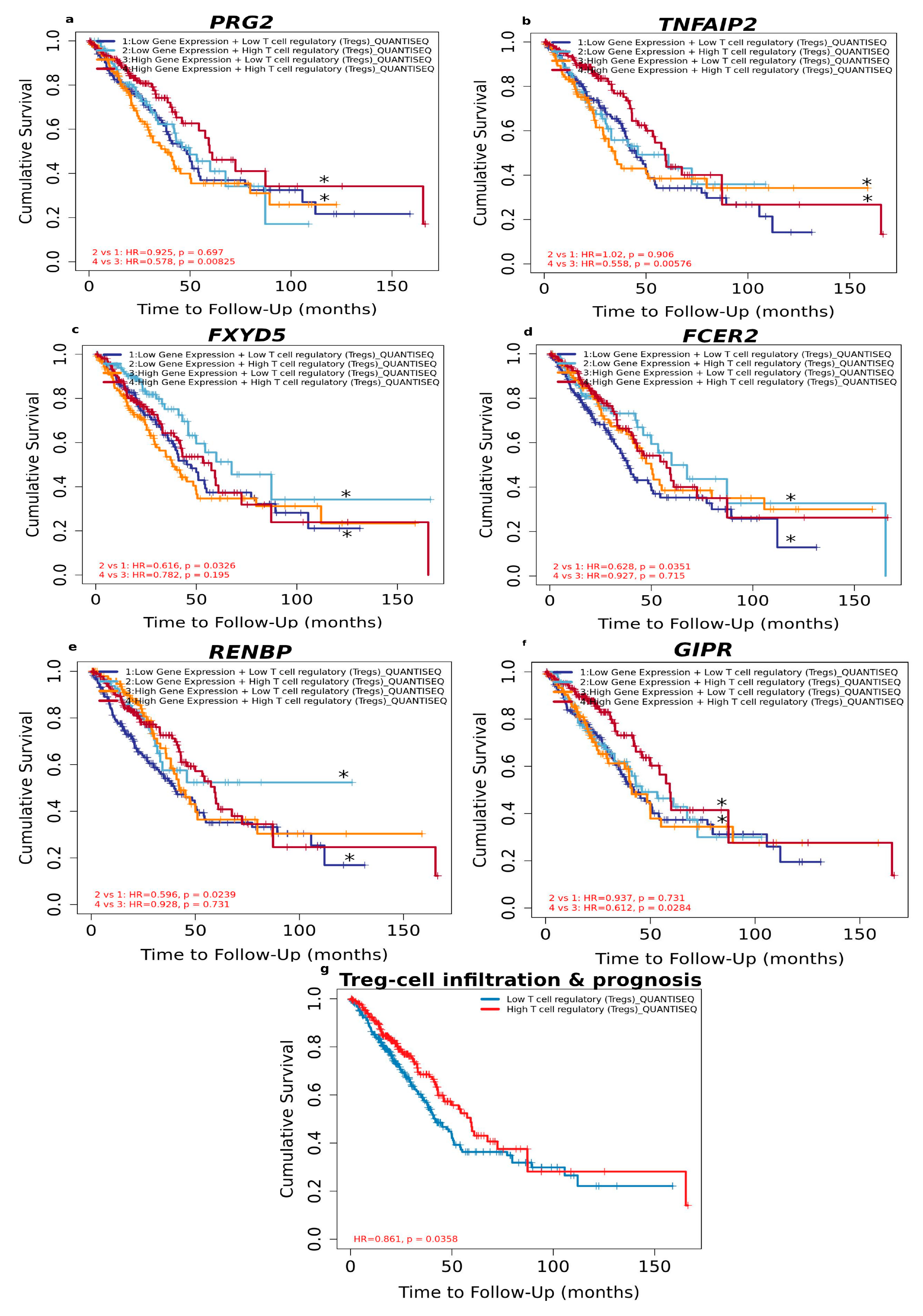
3.1.4. Expression of 22 DEGs Strongly Associates with Tumour Infiltration Abundance of Immune Cells
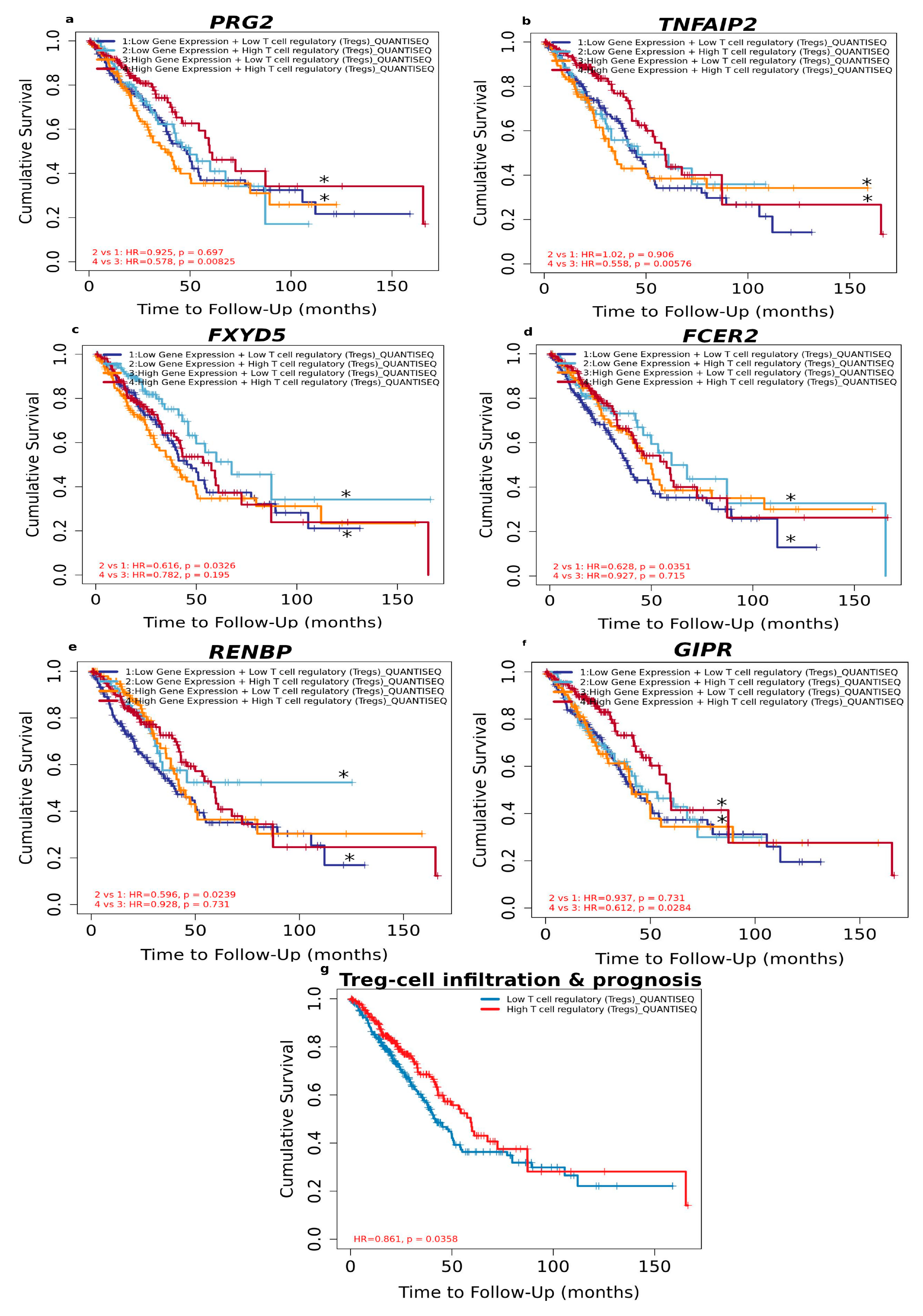
3.1.5. High Level of Infiltrated Regulatory T Cells Accompanied by DEGs Correlates with Better LUAD Prognosis
3.2. Hormone-Related Analyses
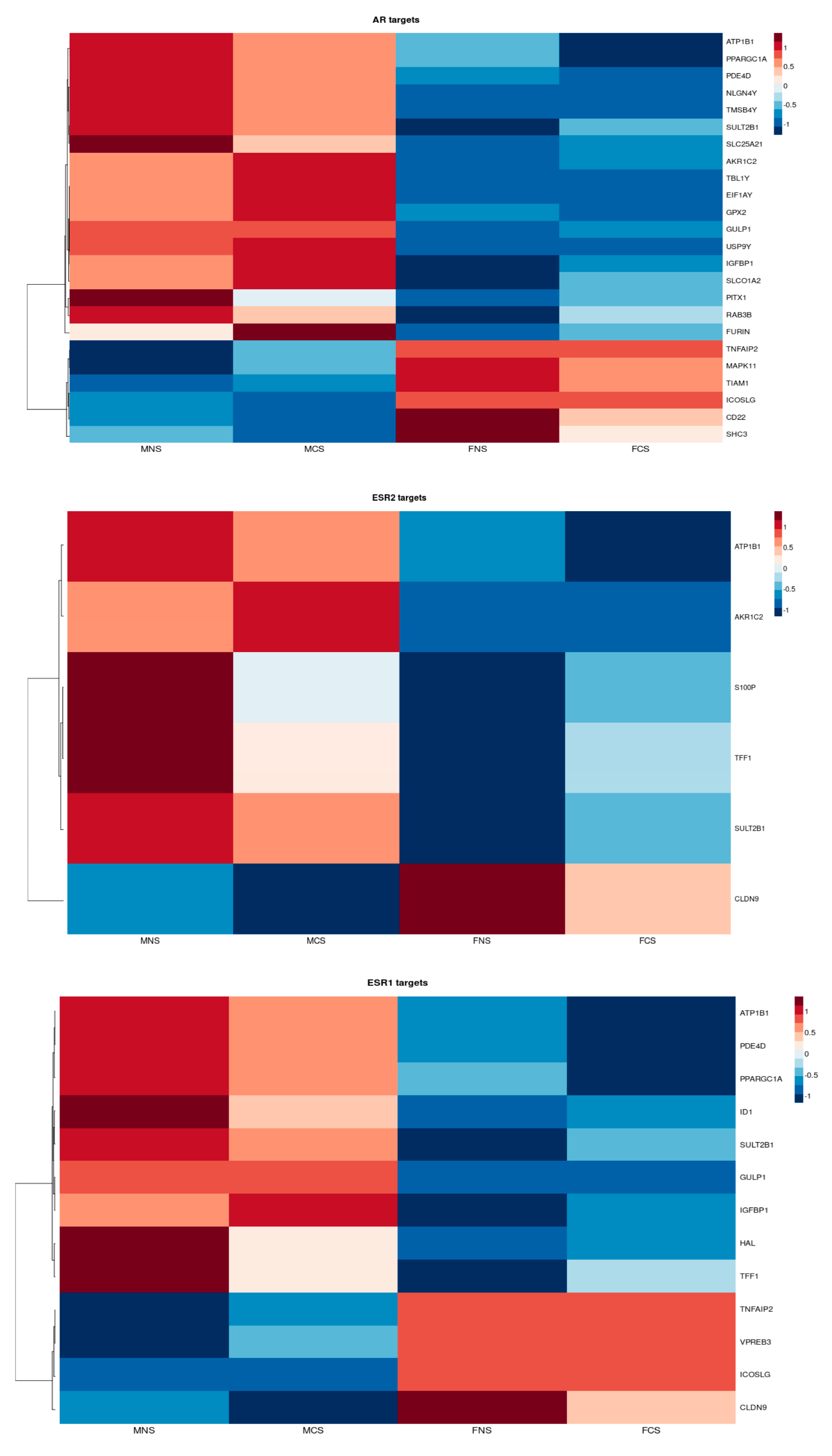
3.2.1. Circulating Hormonal Transcriptional Targets Affect LUAD Tumour Proliferation and Progression
3.2.2. Association of Oestrogen Protection in Males and Premenopausal and Postmenopausal Females
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Barrera, R.; Morales Fuentes, J. Lung cancer in women. Lung Cancer Targets Ther. 2012, 3, 79–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Ramos, C.M.; Quackenbush, J.; DeMeo, D.L. Genome-Wide Sex and Gender Differences in Cancer. Front. Oncol. 2020, 10, 597788. [Google Scholar] [CrossRef] [PubMed]
- Mederos, N.; Friedlaender, A.; Peters, S.; Addeo, A. Gender-specific aspects of epidemiology, molecular genetics and outcome: Lung cancer. ESMO Open 2020, 5, e000796. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; Dacic, S.; Land, S.R.; Lenzner, D.E.; Dhir, R.; Acquafondata, M.; Landreneau, R.J.; Grandis, J.R.; Siegfried, J.M. Combined Analysis of Estrogen Receptor β-1 and Progesterone Receptor Expression Identifies Lung Cancer Patients with Poor Outcome. Clin. Cancer Res. 2011, 17, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Lara, V.; Avila-Costa, M.R. An Overview of Lung Cancer in Women and the Impact of Estrogen in Lung Carcinogenesis and Lung Cancer Treatment. Front. Med. 2021, 8, 600121. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 135–170. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1876162319300112 (accessed on 2 January 2023).
- Mauvais-Jarvis, F. Estrogen and androgen receptors: Regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol. Metab. 2011, 22, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Clocchiatti, A.; Cora, E.; Zhang, Y.; Dotto, G.P. Sexual dimorphism in cancer. Nat. Rev. Cancer 2016, 16, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Hammouz, R.Y.; Kostanek, J.K.; Dudzisz, A.; Witas, P.; Orzechowska, M.; Bednarek, A.K. Differential expression of lung adenocarcinoma transcriptome with signature of tobacco exposure. J. Appl. Genet. 2020, 61, 421–437. [Google Scholar] [CrossRef]
- Home–GEO–NCBI. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 24 September 2022).
- Scott Davis, S. CBDM Bioinformatics. Available online: https://cbdm.hms.harvard.edu/LabMembersPges/SD.html (accessed on 24 September 2022).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Kou, Y.; Chen, E.Y.; Clark, N.R.; Duan, Q.; Tan, C.M.; Ma‘ayan, A. ChEA2: Gene-Set Libraries from ChIP-X Experiments to Decode the Transcription Regulome. In Availability, Reliability, and Security in Information Systems and HCI; Cuzzocrea, A., Kittl, C., Simos, D.E., Weippl, E., Xu, L., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2013; Volume 8127, pp. 416–430. Available online: http://link.springer.com/10.1007/978-3-642-40511-2_30 (accessed on 24 September 2022).
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Wallford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Sopper, S.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Warnes, G.; Bolker, B.; Liaw, W.H.A.; Lumley, T.; et al. gplots: Various R programming tools for plotting data. R Package Version 2009, 2, 1. [Google Scholar]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, M.; Maisonial-Besset, A.; Zhu, Y.; Witkowski, T.; Roche, G.; Boucheix, C.; Greco, C.; Degoul, F. Targeting the Tetraspanins with Monoclonal Antibodies in Oncology: Focus on Tspan8/Co-029. Cancers 2019, 11, 179. [Google Scholar] [CrossRef] [PubMed]
- Guillemette, C.; Bélanger, A.; Lépine, J. Metabolic inactivation of estrogens in breast tissue by UDP-glucuronosyltransferase enzymes: An overview. Breast Cancer Res. 2004, 6, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, J.B.; Lagas, J.S.; Broestl, L.; Sponagel, J.; Rockwell, N.; Rhee, G.; Rosen, S.F.; Chen, S.; Klein, R.S.; Imoukhuede, P.; et al. Sex differences in cancer mechanisms. Biol. Sex Differ. 2020, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheatley-Price, P.; Blackhall, F.; Lee, S.M.; Ma, C.; Ashcroft, L.; Jitlal, M.; Qian, W.; Hackshaw, A.; Rudd, R.; Booton, R.; et al. The influence of sex and histology on outcomes in non-small-cell lung cancer: A pooled analysis of five randomized trials. Ann. Oncol. 2010, 21, 2023–2028. [Google Scholar] [CrossRef]
- Mervic, L. Time Course and Pattern of Metastasis of Cutaneous Melanoma Differ between Men and Women. PLoS ONE 2012, 7, e32955. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, A.; Blenis, J.; Gomes, A.P. Beyond the Warburg Effect: How Do Cancer Cells Regulate One-Carbon Metabolism? Front. Cell Dev. Biol. 2018, 6, 90. [Google Scholar] [CrossRef]
- Liu, J.; Lu, F.; Gong, Y.; Zhao, C.; Pan, Q.; Ballantyne, S.; Zhao, X.; Tian, S.; Chen, H. High expression of synthesis of cytochrome c oxidase 2 and TP53-induced glycolysis and apoptosis regulator can predict poor prognosis in human lung adenocarcinoma. Hum. Pathol. 2018, 77, 54–62. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Sajid, M.; Sample Organization. Immunomodulatory effect of Xenobiotics. J. Environ. Immunol. Toxicol. 2016, 3, 1. [Google Scholar] [CrossRef]
- Nothdurft, S.; Thumser-Henner, C.; Breitenbücher, F.; Okimoto, R.A.; Dorsch, M.; Opitz, C.A.; Sadik, A.; Esser, C.; Hölzel, M.; Asthana, S.; et al. Functional screening identifies aryl hydrocarbon receptor as suppressor of lung cancer metastasis. Oncogenesis 2020, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, X.; Huo, R.; Li, X.; Yang, Y.; Gai, Z.; Xu, M.; Shen, L.; Cai, L.; Wan, C.; et al. Association study of UGT1A9 promoter polymorphisms with DILI based on systematically regional variation screen in Chinese population. Pharm. J. 2015, 15, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Csajka, C.; Dotto, G.P.; Wagner, A.D. Sex Differences in Efficacy and Toxicity of Systemic Treatments: An Undervalued Issue in the Era of Precision Oncology. J. Clin. Oncol. 2018, 36, 2680–2683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, M.; Deng, B.; Dai, N.; Feng, Y.; Shan, J.; Yang, Y.; Mao, C.; Huang, P.; Xu, C.; et al. HLA-DQB1 expression on tumor cells is a novel favorable prognostic factor for relapse in early-stage lung adenocarcinoma. Cancer Manag. Res. 2019, 11, 2605–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Zuo, S.; Wei, M.; Wang, S.; Dong, J.; Wei, J. Pan-Cancer Analysis of Immune Cell Infiltration Identifies a Prognostic Immune-Cell Characteristic Score (ICCS) in Lung Adenocarcinoma. Front. Immunol. 2020, 11, 1218. [Google Scholar] [CrossRef]
- Mafi, S.; Mansoori, B.; Taeb, S.; Sadeghi, H.; Abbasi, R.; Cho, W.C.; Rostamzadeh, D. mTOR-Mediated Regulation of Immune Responses in Cancer and Tumor Microenvironment. Front. Immunol. 2022, 12, 774103. [Google Scholar] [CrossRef]
- Olak, J.; Colson, Y. Gender differences in lung cancer: Have we really come a long way, baby? J. Thorac. Cardiovasc. Surg. 2004, 128, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Brand, J.S.; Chan, M.F.; Dowsett, M.; Folkerd, E.; Wareham, N.J.; Luben, R.N.; Van Der Schouw, Y.T.; Khaw, K.-T. Cigarette Smoking and Endogenous Sex Hormones in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2011, 96, 3184–3192. [Google Scholar] [CrossRef] [Green Version]
- Piipari, R.; Nurminen, T.; Savela, K.; Hirvonen, A.; Mäntylä, T.; Anttila, S. Glutathione S-transferases and aromatic DNA adducts in smokers’ bronchoalveolar macrophages. Lung Cancer 2003, 39, 265–272. [Google Scholar] [CrossRef]
- Barros, R.P.A.; Gustafsson, J.Å. Estrogen Receptors and the Metabolic Network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Cycle | STK11, BIRC7, CROCC,AKR1B10, C19orf21, MST4, PTP4A1 |
| Proliferation | AKR1C2, AKR1C3, ID1, IRS2, PPARGC1A, PTHLH, TFF1,AKR1B10, FOXP2,APOE, BOK, CBFA2T3, CD79A, CX3CL1,ENG, MAPK11, MS4A1, STK11, TNFRSF13B, TNFSF14, ICOSLG, MATK |
| Apoptosis | BOK, STK11, CD14,PPARGC1A, AKR1E2, ATP1B1, MST4,SLCO1A2 |
| PI3K-AKT Signalling Pathway | CDKN1A, EGFR, MTOR, PIK3CD, PIK3R2,CDKN1B, CHUK, IRS2, KIT |
| MAPK signalling pathway | MAP2K1, PRKACB, CHUK, DUSP4, KRAS,MAPK11, CD14,TGFB1, EGFR, MAPK1, MAPK3, NFATC2, RELA, TGFB2, GADD45A, TGFBR2, CACNA1I |
| ATP-dependent transporter of the ATP-binding cassette (ABC) | MGST1, AKR1C3, ALDH3B1, TAT, AKR1C1, AKR1C2,ITIH4, IGFBP1, GSR, ABCC2, PGD, ENPEP, MCCC2,GABARAPL1, GSTM4, GSTO2, GSTA2, PTGES3, AHCY,CYP2C8, CBR1,APOE, PINK1, ALDH1A3, GSTA3, PPARD,TGFB2 |
| HIF PID signalling pathway | ENG, GATA2, FURIN,IGFBP1 |
| FOXO signalling pathway | ARG1, APOE, ATM, CDKN1A, EGFR, GABARAP, GADD45A, MAPK11, MAPK1, MAPK3, PIK3CD, PIK3R2, TGFB1, TGFB2, TGFBR2, STK11,SREBF1, CCNB3, CCND1,CDKN1B, CHUK, GABARAPL1, INSR, IRS2, KRAS,MAP2K1, PRKAB2, PRKAG2, PRMT1, SIRT1, SMAD4, EIF4E, ULK2, FOXP2, PPARGC1A |
| AMPK signalling pathway | CCND1, GYS2, INSR, IRS2, LEP, PPARGC1A, PPP2CB, PPP2R1A, PPP2R1B, PPP2R5C, PRKAB2, PRKAG2, SIRT1, SREBF1,STK11, CCNA1, PPP2R5B, CREB3, PFKL, PIK3CD,PIK3R2, RAB8A, STK11, STRADB, TBC1D1 |
| mTOR signalling pathway | EIF4B, INSR, SEH1L, CHUK, MAP2K1, ATP6V1G1, EIF4E,ULK2, PTHLH,STK11, MAPK1, PIK3R2, WNT6, ATP6V1E1, FZD7, STK11, TELO2, MAPK3, WNT5B, FZD2,MTOR, FLCN, PIK3CD, WDR24, NPRL2, STRADB, PIK3CG |
| TIMER2.0 immune related genes | RENBP, ARHGAP22, CD79A, CXCR5, FCER2, GIPR,MS4A1, NCR3, PRG2, RYR1, SLC15A3, TNFRSF13B, TNFSF14, VPREB3, FGD3, FXYD5, HLA-DQB1, HMHA1,ICOSLG, TMC8, TNFAIP2,HAL |
| Warburg Effect | ACSS1, PFKL, PIK3CD, GLS, EGFR, HK1, MAPK1, PGAM2,MAPK3, PIK3R2,PDHB, KIT, KRAS, G6PD, MAP2K1S |
| ARH signalling pathway | CDKN1A, EGFR, RELA, TGFB1, MAPK1,GSTA2, IGFBP1,KRAS, MAP2K1, CDKN1B |
| Metabolism of xenobiotics by cytochrome P450 | CYP2C8, MGST1, AKR1C3, ALDH3B1, AKR1C2, AKR1C1,GSTM4, GSTO2, GSTA2,ALDH1A3, CYP2S1, GSTA3 |
| RAS signalling pathway | APOE, MAPK11, RTN4R, SHC3, STMN3, TIAM1, SHC2 |
| Wnt signalling pathway | NFATC3, WNT6, FZD7,TBL1Y, NFATC2, CCND1, WNT5B,PRKACB, FZD2 |
| Reactive Oxygen Species pathway | ABCC1, G6PD, GLRX2, GSR, MGST1, NDUFB4, PRDX4,PRDX6, TXNRD2 |
| DEGs | Purity | B Lymphocytes | CD8+ T cell | CD4+ T cell | Macrophages | NEUTROPHILES | DC | Tregs | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 | P | R2 | P | R2 | P | R2 | P | R2 | P | R2 | P | R2 | P | R2 | P | |
| ARHGAP22 | −0.229 | 2.65 × 10−7 | 0.175 | 9.26 × 10−5 | 0.063 | 1.63 × 10−1 | 0−0.204 | 5.07 × 10−6 | 0.1 | 2.65 × 10−2 | 0.045 | 3.22 × 10−1 | 0.141 | 1.66 × 10−3 | 0.189 | 2.37 × 10−5 |
| CD79A | −0.457 | 7.17 × 10−27 | 0.718 | 1.89 × 10−79 | 0.403 | 1.06 × 10−20 | −0.161 | 3.36 × 10−4 | −0.013 | 7.79 × 10−1 | −0.142 | 1.56 × 10−3 | 0.071 | 1.15 × 10−1 | 0.482 | 4.26 × 10−30 |
| CXCR5 | −0.505 | 2.24 × 10−33 | 0.731 | 1.66 × 10−83 | 0.45 | 6.43 × 10−26 | −0.115 | 1.03 × 10−2 | 0.135 | 2.69 × 10−3 | −0.025 | 5.84 × 10−1 | −0.004 | 9.27 × 10−1 | 0.501 | 1.04 × 10−32 |
| FCER2 | −0.424 | 5.88 × 10−23 | 0.58 | 1.33 × 10−45 | 0.271 | 1.01 × 10−9 | −0.022 | 6.23 × 10−1 | 0.104 | 2.06 × 10−2 | 0.004 | 9.34 × 10−1 | −0.073 | 1.06 × 10−1 | 0.376 | 5.14 × 10−18 |
| FGD3 | −0.423 | 7.46 × 10−23 | 0.455 | 1.58 × 10−26 | 0.498 | 3.08 × 10−32 | −0.215 | 1.52 × 10−6 | 0.282 | 1.72 × 10−10 | 0.02 | 6.65 × 10−1 | 0.009 | 8.40 × 10−1 | 0.516 | 7.64 × 10−35 |
| FXYD5 | −0.267 | 1.60 × 10−9 | 0.031 | 4.89 × 10−1 | 0.06 | 1.83 × 10−1 | −0.026 | 5.70 × 10−1 | 0.328 | 8.45 × 10−14 | 0.233 | 1.77 × 10−7 | −0.198 | 9.33 × 10−6 | 0.069 | 1.28 × 10−1 |
| GIPR | −0.104 | 2.12 × 10−2 | 0.21 | 2.61 × 10−6 | 0.044 | 3.20 × 10−1 | −0.148 | 9.68 × 10−4 | 0.205 | 4.46 × 10−6 | 0.171 | 1.39 × 10−4 | −0.079 | 7.89 × 10−2 | 0.362 | 1.09 × 10−16 |
| HAL | 0.047 | 2.90 × 10−1 | −0.063 | 1.59 × 10−1 | −0.158 | 4.23 × 10−4 | 0.129 | 4.04 × 10−13 | −0.191 | 1.93 × 10−5 | 0.071 | 1.16 × 10−1 | 0.08 | 7.64 × 10−2 | −0.122 | 6.48 × 10−3 |
| HLA−DQB1 | −0.353 | 6.30 × 10−16 | 0.247 | 2.90 × 10−8 | 0.226 | 3.87 × 10−7 | −0.126 | 4.99 × 10−3 | 0.445 | 2.51 × 10−25 | 0.1 | 2.70 × 10−2 | −0.204 | 4.82 × 10−6 | 0.342 | 5.78 × 10−15 |
| HMHA1 | −0.277 | 3.97 × 10−10 | 0.218 | 1.06 × 10−6 | 0.21 | 2.54 × 10−6 | −0.234 | 1.39 × 10−7 | 0.359 | 2.02 × 10−16 | 0.063 | 1.65 × 10−1 | −0.056 | 2.13 × 10−1 | 0.408 | 3.49 × 10−21 |
| ICOSLG | −0.233 | 1.59 × 10−7 | 0.3 | 1.06 × 10−11 | 0.12 | 7.55 × 10−3 | −0.042 | 3.49 × 10−1 | 0.181 | 5.51 × 10−5 | 0.038 | 3.97 × 10−1 | 0.023 | 6.11 × 10−1 | 0.401 | 1.80 × 10−20 |
| MS4A1 | −0.502 | 7.29 × 10−33 | 0.74 | 1.19 × 10−86 | 0.441 | 7.14 × 10−25 | −0.086 | 5.76 × 10−2 | 0.082 | 6.93 × 10−2 | −0.068 | 1.30 × 10−1 | −0.028 | 5.32 × 10−1 | 0.454 | 2.22 × 10−26 |
| NCR3 | −0.503 | 4.88 × 10−33 | 0.607 | 5.62 × 10−51 | 0.63 | 5.46 × 10−56 | −0.101 | 2.46 × 10−2 | 0.155 | 5.68 × 10−4 | −0.092 | 4.01 × 10−2 | −0.079 | 7.94 × 10−2 | 0.431 | 9.64 × 10−24 |
| PRG2 | −0.122 | 6.43 × 10−3 | −0.022 | 6.32 × 10−1 | −0.061 | 1.76 × 10−1 | −0.236 | 1.11 × 10−7 | 0.25 | 1.95 × 10−8 | 0.197 | 1.06 × 10−5 | −0.085 | 5.97 × 10−2 | 0.179 | 6.57 × 10−5 |
| RENBP | −0.237 | 9.51 × 10−8 | 0.173 | 1.18 × 10−4 | 0.236 | 1.16 × 10−7 | −0.178 | 7.16 × 10−5 | 0.268 | 1.42 × 10−9 | 0.071 | 1.16 × 10−1 | 0.01 | 8.28 × 10−1 | 0.265 | 2.28 × 10−9 |
| RYR1 | −0.112 | 1.31 × 10−2 | 0.05 | 2.63 × 10−1 | 0.092 | 4.18 × 10−2 | −0.113 | 1.20 × 10−2 | 0.341 | 6.33 × 10−15 | 0.234 | 1.47 × 10−7 | −0.094 | 3.68 × 10−2 | 0.203 | 5.50 × 10−6 |
| SLC15A3 | −0.431 | 9.65 × 10−24 | 0.269 | 1.23 × 10−9 | 0.345 | 2.90 × 10−15 | −0.132 | 3.35 × 10−3 | 0.383 | 1.16 × 10−18 | 0.071 | 1.18 × 10−1 | −0.107 | 1.76 × 10−2 | 0.461 | 2.38 × 10−27 |
| TMC8 | −0.432 | 8.18 × 10−23 | 0.404 | 8.79 × 10−21 | 0.406 | 5.60 × 10−21 | −0.169 | 1.68 × 10−4 | 0.344 | 4.07 × 10−15 | 0.071 | 1.18 × 10−1 | −0.19 | 2.10 × 10−5 | 0.472 | 1.17 × 10−28 |
| TNFAIP2 | −0.311 | 1.51 × 10−12 | 0.167 | 2.01 × 10−4 | 0.3 | 1.03 × 10−11 | −0.19 | 2.25 × 10−5 | 0.312 | 1.44 × 10−12 | 0.041 | 3.59 × 10−1 | −0.029 | 5.26 × 10−1 | 0.275 | 5.45 × 10−10 |
| TNFRSF13B | −0.438 | 1.37 × 10−24 | 0.717 | 3.98 × 10−79 | 0.365 | 5.76 × 10−17 | −0.096 | 3.23 × 10−2 | 0.041 | 3.69 × 10−1 | −0.039 | 3.84 × 10−1 | −0.066 | 1.46 × 10−1 | 0.478 | 1.79 × 10−29 |
| TNFSF14 | −0.356 | 3.34 × 10−16 | 0.21 | 2.67 × 10−6 | 0.206 | 4.18 × 10−6 | 0.292 | 3.95 × 10−11 | 0.181 | 6.01 × 10−5 | 0.045 | 3.22 × 10−1 | −0.109 | 1.52 × 10−2 | 0.264 | 2.70 × 10−9 |
| VPREB3 | −0.442 | 5.33 × 10−25 | 0.688 | 2.18 × 10−70 | 0.308 | 2.53 × 10−12 | −0.116 | 1.01 × 10−2 | −0.042 | 3.47 × 10−1 | −0.132 | 3.26 × 10−3 | 0.042 | 3.50 × 10−1 | 0.284 | 1.34 × 10−10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammouz, R.Y.; Orzechowska, M.; Anusewicz, D.; Bednarek, A.K. X or Y Cancer: An Extensive Analysis of Sex Differences in Lung Adenocarcinoma. Curr. Oncol. 2023, 30, 1395-1415. https://doi.org/10.3390/curroncol30020107
Hammouz RY, Orzechowska M, Anusewicz D, Bednarek AK. X or Y Cancer: An Extensive Analysis of Sex Differences in Lung Adenocarcinoma. Current Oncology. 2023; 30(2):1395-1415. https://doi.org/10.3390/curroncol30020107
Chicago/Turabian StyleHammouz, Raneem Yaseen, Magdalena Orzechowska, Dorota Anusewicz, and Andrzej K. Bednarek. 2023. "X or Y Cancer: An Extensive Analysis of Sex Differences in Lung Adenocarcinoma" Current Oncology 30, no. 2: 1395-1415. https://doi.org/10.3390/curroncol30020107