Braf-Mutant Melanomas: Biology and Therapy



Abstract
:1. Introduction
2. BRAF-Mutated Melanomas
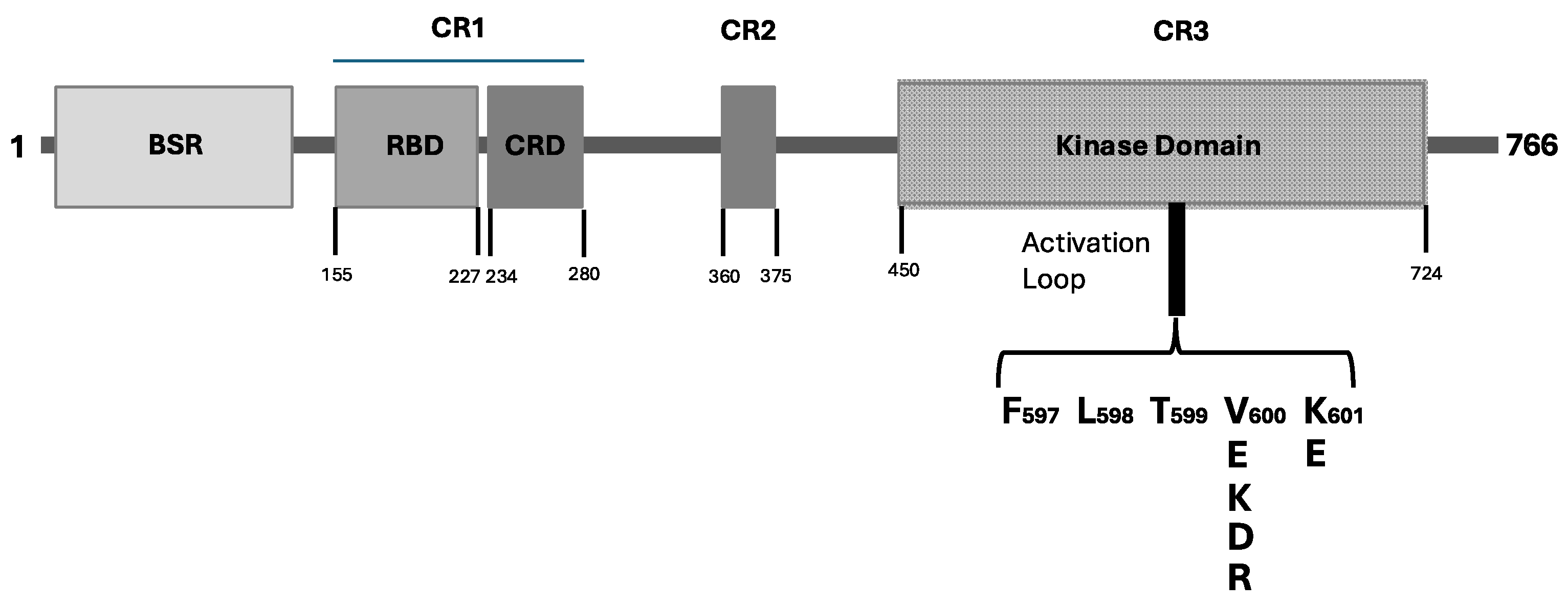
2.1. BRAF Mutations
2.2. Role of BRAF Mutations in Melanomagenesis
3. Therapy of BRAF-Mutant Melanomas
3.1. Adjuvant Therapy in Stage II Melanoma
3.2. Adjuvant Therapy in Stage III Melanoma
3.3. Neoadjuvant Therapy of Melanoma
3.4. Adjuvant Vaccination Studies
3.5. Immunotherapy and Targeted Therapy of Metastatic Melanoma
3.6. Adoptive Therapy with Tumor-Infiltrating T Lymphocytes (TILs) in Melanoma Patients Who Have Failed Immunotherapy and/or Targeted Therapy Treatments
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Elder, D.E.; Bastian, B.C.; Cree, J.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization classification of cutaneous, mucosal and uveal melanoma. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Castelli, G.; Pelosi, E. Melanoma genetic abnormalities, tumor progression, clonal evolution and tumor initiating cells. Med. Sci. 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Yong, T.T.; Yu, S.; Khale Ke, C.L.; Cheng, S.T. The genomic landscape of melanoma and its therapeutic implications. Genes 2023, 14, 1021. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.; áArman Aksoy, B.; Albert, M.; Ally, A.; Amin, S.; Arachchi, H.; Arora, A.; áTodd Auman, J.; Ayala, B.; et al. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Druskovich, C.; Kelley, J.; Aubrey, J.; Palladino, L.; Wright, G.P. A review of melanoma subtypes: Genetic and treatment considerations. J. Surg. Oncol. 2024, in press. [Google Scholar] [CrossRef]
- Salhi, A.; Farhadian, J.A.; Giles, K.M.; Vega-Saenz de Miera, E.; Silva, I.P.; Bourque, C.; Yeh, K.; Chhangalawa, S.; Wang, J.; Ye, F.; et al. RSK1 activation promotes invasion in nodular melanoma. Am. J. Pathol. 2015, 185, 704–710. [Google Scholar] [CrossRef]
- Trebino, T.E.; Markusic, B.; Nan, H.; Banerjee, S.; Wang, Z. Unveiling the domain-specific and Ras isoform-specific details of BRAF kinase regulation. eLife 2023, 12, RP88836. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Gonzalez-del Pino, G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF-MEK-14-3-3 complex. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef]
- Martinez-Fiesco, J.A.; Durrant, D.E.; Morrison, D.K.; Zhang, P. Structural insights into the BRAF monomer to dimer transition mediated by RAS binding. Nat. Commun. 2022, 13, 486. [Google Scholar] [CrossRef]
- Kazandjian, S.; Rousselle, E.; Danker, M.; Cescon, D.W.; Spreafico, A.; Ma, K.; Kavan, P.; Batist, G.; Rosae, A. The clinical, genomic, and transcriptomic landscape of BRAF mutant cancers. Cancers 2024, 16, 445. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Nepote, A.; Avallone, G.; Ribero, S.; Cavallo, F.; Roccuzzo, G.; Mastorino, L.; Conforti, C.; Paruzzo, L.; Poletto, S.; Carnevale Schienca, F. Current controversies and challenges on BRAF V660K-mutant cutaneous melanoma. J. Clin. Med. 2022, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Zengarini, C.; Mussi, M.; Veronesi, G.; Alessandrini, A.; Lambertini, M.; Dika, E. BRAF V600K vs. BRAF V600E: A comparison of clinical and dermoscopic characteristics and response to immunotherapies and targeted therapies. Clin. Exp. Dermatol. 2022, 47, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Nazzaro, G.; Maronese, C.A.; Casazza, G.; Giacalone, S.; Spigariolo, C.B.; Roccuzzo, G.; Avallone, G.; Guida, S.; Brancaccio, G.; Broganelli, P.; et al. Dermoscopic predictors of melanoma in small diameter melanocytic lesions (Mini-melanoma): A retrospective multicentric study of 269 cases. Int. J. Dermatol. 2023, 62, 1040–1049. [Google Scholar] [CrossRef]
- Hanrahan, A.J.; Chen, Z.; Rosen, N.; Solit, D.B. BRAF—A tumor-agnostic drug target with lineage-specific dependencies. Nat. Rev. Clin. Oncol. 2024, 21, 224–247. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Dahlman, K.B. Class matters: Sensitivity of BRAF-mutant melanoma to MAPK inhibition. Clin. Cancer Res. 2018, 24, 6107–6109. [Google Scholar] [CrossRef]
- Dankner, M.; Lajoie, M.; Moldoveanu, D.; Nguyen, T.T.; Savage, P.; Rajkumar, S.; Huang, X.; Lvova, M.; Protopopov, A.; Vuzmann, D.; et al. Dual MAPK inhibition is an effective therapeutic strategy for a subset of class II BRAF mutant melanomas. Clin. Cancer Res. 2018, 24, 6483–6494. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Wang, Y.; Fazeldad, R.; Johnson, B.; Nebhan, C.A.; Dagogo-Jack, I.; Myall, M.J.; Richtig, G.; Bracht, J.; Gerlinger, M.; et al. Clinical activity of mitogen-activated protein kinase-targeted therapies in patients with non-V600 BRAF-mutant tumors. JCO Precis. Oncol. 2022, 6, e2200107. [Google Scholar] [CrossRef]
- Turner, J.A.; Bemis, J.; Bagby, S.; Capasso, A.; Yacob, B.; Chimed, T.S.; van Gulick, R.; Lee, H.; Tobin, R.; Tentler, J.J.; et al. BRAF fusions identified in melanomas have variable treatment responses and phenotypes. Oncogene 2019, 38, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Botton, T.; Televich, E.; Mishra, V.; Zhang, T.; Shain, A.H.; Berquel, C.; Gagnon, A.; Judson, R.L.; Ballotti, R.; Ribas, A. Genetic heterogeneity of BRAF fusion kinases in melanoma affects drug responses. Cell Rep. 2019, 29, 573–588. [Google Scholar] [CrossRef]
- Moran, J.; Le, L.; Nardi, V.; Golas, J.; Farahani, A.; Signorelli, S.; Onozato, M.L.; Foreman, R.K.; Duncan, L.; Lawrence, D.P.; et al. Identification of fusions with potential clinical significance in melanoma. Mod. Pathol. 2022, 35, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Birkealv, S.; Harland, M.; Matuyama, L.; Rashid, M.; Mehta, I.; Laye, J.P.; Haase, K.; Mell, T.; Iyer, V.; Robles-Espinoza, C.D.; et al. Mutually exclusive genetic interactions and gene essentiality shape the genomic landscape of primary melanoma. J. Pathol. 2023, 259, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.; Berry, D.; Heney, K.A.; Strong, C.; Ramsay, L.A.; Lajoie, M.; Alkallas, R.; Nguyen, T.T.; Thomson, C.; Adams, M.A.; et al. Melanomas with concurrent BRAF non-p.V600 and NF1 loss-of-function mutations are targeatable by BRAF/MEK inhibitor combination therapy. Cell Rep. 2022, 39, 110634. [Google Scholar] [CrossRef]
- Bastian, B.C.; Le Boit, P.E.; Hamm, H.; Brocker, E.B.; Pinkel, D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998, 58, 2170–2175. [Google Scholar] [PubMed]
- Maldonado, J.L.; Fridlyand, J.; Patel, H.; Jain, A.N.; Busam, K.; Kageshita, T.; Ono, T.; Albertson, D.G.; Pinkel, D.; Bastian, B.C. Determinants of BRAF mutations in primary melanomas. J. Natl. Cancer Inst. 2003, 95, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Hélias-Rodzewicz, Z.; Frunck-Brentano, E.; Baudoux, L.; Jung, C.K.; Zimmermann, U.; Marin, C.; Clerici, T.; Le Gall, C.; Peschaud, F.; Taly, V.; et al. Variations in BRAF mutant allele percentage in melanomas. BMC Cancer 2015, 15, 497. [Google Scholar] [CrossRef]
- Stagni, C.; Zamuner, C.; Elefanti, L.; Zanin, T.; del Bianco, P.; Sommariva, A.; Fabozzi, A.; Pigozzo, J.; Mocellin, S.; Montesco, M.C.; et al. BRAF gene copy number and mutant allele frequency correlate with time to progression in metastatic melanoma patients treated with MAPK inhibitors. Mol. Cancer Ther. 2018, 17, 1332–1340. [Google Scholar] [CrossRef]
- Birkeland, E.; Zhang, S.; Poduval, D.; Geisler, J.; Nakken, S.; Vodak, D.; Meza-Zepeda, L.A.; Hovig, E.; Myklebost, O.; Knappskog, S.; et al. Patterns of genomic evolution in advanced melanoma. Nat. Commun. 2018, 9, 2665. [Google Scholar] [CrossRef]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef]
- Colebatch, A.J.; Ferguson, P.; Newell, F.; Kazakoff, S.H.; Witkowski, T.; Dobrovic, A.; Johansson, P.A.; Saw, R.; Strecth, J.R.; McArthur, G.A.; et al. Molecular genomic profiling of melanocytic nevi. J. Investig. Dermatol. 2019, 139, 1762–1768. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.; von Deimling, A.; Bastian, B.C. Clonal BRAF mutations in melanocytic nevi and initiating role of BRAF in melanocytic neoplasia. J. Natl. Cancer Inst. 2013, 105, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Widlund, H.R.; Kutok, J.L.; Kopanl, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.; et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr. Biol. 2005, 15, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Tavelich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Joseph, N.M.; Yu, R.; Benhamida, J.; Liu, S.; Prow, T.; Ruben, B.; North, J.; Pincus, L.; Yeh, I.; et al. Genomic and transcriptomic analysis reveals incrementsl disruption of key signaling pathways during melanoma evolution. Cancer Cell 2018, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Matsuta, M.; Imamura, Y.; Matsuta, M.; Sasaki, K.; Kon, S. Detection of numerical chromosomal aberrations in malignant melanomas using fluorescence in situ hybridization. J. Cutan. Pathol. 1997, 24, 201–205. [Google Scholar] [CrossRef]
- Casorzo, L.; Luzzi, C.; Nardacchione, A.; Picciotto, F.; Pisacana, A.; Risio, M. Fluorescence in situ hybridization (FISH) evaluation of chromosome 6, 7, 9 and 10 throughout human melanocytic tumorigenesis. Melanoma Res. 2005, 15, 155–160. [Google Scholar] [CrossRef]
- Udart, M.; Utikal, J.; Krahn, G.M.; Peter, R.U. Chromosome 7 aneusomy. A marker for metastatic melanomas? Expression of the epidermal growth factor receptor gene and chromosome 7 aneusomy in nevi, primary malignant melanomas and metastases. Neoplasia 2001, 3, 245–254. [Google Scholar] [CrossRef]
- Luke, J.J.; Ascierto, P.A.; Khattak, M.A.; de la Cruz Merino, L.; del Vecchio, M.; Rutkowski, P.; Spagnolo, F.; Machiewicz, J.; Chiarion-Sileni, V.; Kirkwood, J.M.; et al. Pembrolizumab versus placebo as adjuvant therapy in resected stage IIB or IIC melanoma: Final analysis of distant metastasis-free survival in the phase III KEYNOTE-716 study. J. Clin. Oncol. 2024, 42, 1619–1624. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; del Vecchio, M.; Weber, J.; Hoeller, C.; Grob, J.J.; Mohr, P.; Loquai, C.; Dutriaux, C.; Chiarion-Sileni, V.; Machiewicz, J.; et al. Adjuvant nivolumab in resected stage IIB/C melanoma: Primary results from the randomized, phase 3 CHECK Mate 76K trial. Nat. Med. 2023, 20, 2835–2843. [Google Scholar] [CrossRef]
- Akkoi, A.; Hauschild, A.; Long, G.; Mandala, M.; Kicinski, M.; Govaerts, A.S.; Klauck, I.; Ouali, M.; Longan, P.C.; Eggermont, A. COLUMBUS-AD: A phase III study of adjuvant encorafenib+binimetinib in resected stage IIB/IIC BRAF V600-mutated melanoma. Future Oncol. 2023, 19, 2017–2027. [Google Scholar] [CrossRef]
- Lee, R.; Rothwell, D.G.; Jackson, R.; Smith, N.; Wong, S.Q.; Kelso, N.; Burghel, G.; Hewitt, C.; Clarke, H.; Michell, J.; et al. DETECTION phase II/III trial: Circulating tumor DNA-guided therapy for stage IIB/C melanoma after surgical resection. J. Clin. Oncol. 2022, 40 (Suppl. S16), TPS9603. [Google Scholar] [CrossRef]
- Tan, L.; Sandhu, S.; Lee, R.J.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann. Oncol. 2019, 30, m804–m814. [Google Scholar] [CrossRef]
- Lee, R.J.; Gremel, G.; Marshall, A.; Myers, K.A.; Fishjer, N.; Dunn, J.A.; Dhomen, N.; Corrie, P.G.; Middleton, M.R.; Loriogan, P.; et al. Circulating tumor DNA predicts xurvival in patients with resected high-risk stage II/III melanoma. Ann. Oncol. 2018, 29, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Brunsgaard, E.K.; Kuzel, T.; Tan, A.; Rush Medical College; Rush University Medical Center. Detection of molecular residual disease in stage II and III melanoma utilizing circulating tumor DNA. J. Clin. Oncol. 2024, 42 (Suppl. S16), e21564. [Google Scholar] [CrossRef]
- Kolliapra, R.; Budde, G.; Aushev, V.N.; Brunsgaard, E.K.; Kuzel, T.; O’Donoughe, C.; Riddell, T.; Palsuledesai, C.C.; Krainock, M.; Liu, M.C.; et al. Logitudinal circulating tumor DNA monitoring for detection of molecular residual disease in patients with surgically resected stage II/III melanoma. J. Clin. Oncol. 2023, 40 (Suppl. S16), 9582. [Google Scholar] [CrossRef]
- Eggermont, A.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomized, double-blind, phase I trial. Lancet Oncol. 2015, 16, 522–530. [Google Scholar] [CrossRef]
- Eggermont, A.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of stage III melanoma: Long-term follow-up results of the European Organization for Research and Treatment of Cancer 18071 double-blind phase 3 randomised trial. Eur. J. Cancer 2019, 119, 1–10. [Google Scholar] [CrossRef]
- Eggermont, A.; Kicinski, M.; Blank, C.; Mandala, M.; Long, G.; Atkinson, V.; Dalle, S.; Haydon, A.; Mechcheryakov, A.; Khattak, A.; et al. Five-year analysis of adjuvant pembrolizumab or placebo in stage III melanoma. N. Engl. J. Med. Evid. 2022, 1, EVIDoa2200214. [Google Scholar] [CrossRef]
- Eggermont, A.; Kicinski, M.; Blank, C.; Mandala, M.; Long, G.; Atkinson, V.; Dalle, S.; Haydon, A.; Mechcheryakov, A.; Khattak, A.; et al. Seven-year analysis of adjuvant pembrolizumab versus placebo in stage III melanoma in the EORTC1325/KEYNOTE-054 trial. Eur. J. Cancer 2024, 211, 114327. [Google Scholar] [CrossRef]
- Weber, J.; del Vecchio, H.J.; Mandala, M.; Gogas, A.M.; Arance, C.L.; Cowey, C.L.; Dalle, S.; Schenkler, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Mandala, M.; del Vecchio, H.J.; Gogas, A.M.; Arance, C.L.; Cowey, C.L.; Dalle, S.; Schenkler, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Outcomes with postrecurrence systemic therapy following adjuvant checkpoint inhibitor treatment for resected melanoma in Check Mate 238. J. Clin. Oncol. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; del Vecchio, M.; Mandal, M.; Gogas, H.; Arance Fernandez, A.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab versus ipilimumab in resected stage III/IV melanoma: 5-year efficacy and biomarker results from CheckMate 238. Clin. Cancer Res. 2023, 29, 3352–3361. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K.F.; Othus, M.; Patel, S.P.; Trhini, A.A.; Sondak, V.K.; Knopp, M.V.; Petrella, T.M.; Truong, T.G.; Khushalani, N.; Cohen, J.V.; et al. Adjuvant pembrolizumab versus IFNα2b or ipilimumab in resected high-risk melanoma. Cancer Discov. 2022, 12, 644–653. [Google Scholar] [CrossRef]
- Linger, J.M.; Darke, A.; Othus, M.; Truong, T.G.; Kushalani, N.; Kendra, K.; Lewis, K.D.; Faller, B.; Funchain, P.; Buchbinder, E.I.; et al. Effectiveness of adjuvant pembrolizumab vs. high-dose interferon or ipilimumab for quality-of-life outcomes in patients with resected melanoma: Secondary analysis of the SWOG S1404 randomized clinical trial. JAMA Oncol. 2023, 9, 251–260. [Google Scholar]
- Livingstone, E.; Zimmer, L.; Hassel, J.C.; Fluck, M.; Eigentler, T.K.; Loquai, C.; Haferkamp, S.; Haferkamp, S.; Gutzmer, R.; Meier, F.; et al. Adjuvant nivolumab plus ipilimumab or nivolumab alone versus placebo in patients with resected stage IV melanoma with no evidence of disease: Final results of a randomized, double-blind, phase 2 trial. Lancet 2022, 400, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Schadedndorf, D.; del Vecchio, M.; Larkin, J.; Atkinson, V.; Schenker, M.; Pigozzo, J.; Gogas, H.; Dalle, S.; Meyer, N.; et al. Adjuvant therapy of nivolumab combined with ipilimumab versus nivolumab alone in patients with resected stage IIIB-D or stage IV melanoma (CheckMate 915). J. Clin. Oncol. 2023, 41, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Hauschild, A.; Santinomi, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 2017, 377, 3813–3823. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Hauschild, A.; Santinami, M.; Kirkwood, J.M.; Atkinson, V.; Mandalà, M.; Merelli, B.; Chiarion-Sileni, V.; Nyakas, M.; Haydon, A.; et al. Final results for adjuvant dabrafenib plus trametinib in stage III melanoma. N. Engl. J. Med. 2024, 391, 1709–1720. [Google Scholar] [CrossRef]
- Dummer, R.; Brase, J.C.; Garett, J.; Campbell, C.D.; Gasal, E.; Squires, M.; Gusenleitener, D.; Santinami, M.; Atkinson, V.; Mandalà, M.; et al. Adjuvant dabrafenib plus trametinib versus placebo in patients with resected, BRAFV600-mutant, stage III melanoma (COMBI-AD): Exploratory biomarker analyses from a randomized, phase 3 trial. Lancet Oncol. 2020, 21, 358–372. [Google Scholar] [CrossRef]
- Grover, P.; Li, I.; Kuijpers, A.; Kreidieh, F.Y.; Williamson, A.; Amaral, T.A.M.; Dimitriou, F.; Placzke, J.; Olino, K.; Vitale, M.G.; et al. Efficacy of adjuvant therapy in patients (pts) with AJCC v8 stage IIIA cutaneous melanoma. J. Clin. Oncol. 2023, 142 (Suppl. S16), 9518. [Google Scholar] [CrossRef]
- Bloem, M.; de Meza, M.M.; Aarts, M.; van den Berkmortel, F.; Blank, C.; Blokx, W.; Boers-Sondersen, M.; Bonenkamp, H.J.; de Groot, J.W.; Haanen, J.; et al. Ajuvant BRAF/MEK versus anti-PD-1 in BRAF-mutant melanoma: Propensity score-matched recurrence-free, distant metastasis-free, an overall survival. J. Clin. Oncol. 2023, 42 (Suppl. S16), 9573. [Google Scholar] [CrossRef]
- Bai, X.; Shaheen, A.; Grieco, C.; D’Arienzo, P.; Mina, F.; Czapla, J.; Lawless, A.; Bongiovanni, E.; Santaniello, U.; Zappi, H.; et al. Dabrafenib plus trametinib versus anti-PD-1 monotherapy ad adjuvant therapy in BRAF V600-mutant stage III melanoma after definitive surgery: A multicenter, retrospective cohort study. Lancet 2023, 65, 102290. [Google Scholar]
- Lodde, G.C.; Hassel, J.; Wulfken, L.M.; Meier, F.; Mohr, P.; Kahler, K.; Hauschild, A.; Schilling, B.; Loquai, C.; Berking, C.; et al. Adjuvant treatment and outcome of stage III melanoma patients: Results of a multicenter real-world German Dermatologic Cooperative Oncology Group (DeCOG) study. Eur. J. Cancer 2023, 191, 112957. [Google Scholar] [CrossRef] [PubMed]
- Roccuzzo, G.; Fava, P.; Astrua, C.; Brizio, M.G.; Cavaliere, G.; Bongiovanni, E.; Sntaniello, U.; Carpentieri, G.; Cangiolosi, L.; Brondino, C.; et al. Real-life outcomes of adjuvant targeted therapy and anti-PD1 agents in stage III/IV resected melanoma. Cancers 2024, 16, 3095. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.N.; Shoushtari, A.N.; Chauhan, D.; Palmieri, D.J.; Lee, B.; Rohaan, M.W.; Mangana, J.; Atkinson, V.; Zaman, F.; Young, A.; et al. Management of early melanoma recurrence despite adjuvant anti-PD1 antibody therapy. Ann. Oncol. 2020, 31, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Bhave, P.; Pallan, L.; Long, G.V.; Menzies, A.M.; Atkinson, V.; Cohen, J.V.; Sullivan, R.J.; Chiarion-Sileni, V.; Nyakas, M.; Khler, K.; et al. Melanoma recurrence patterns and management after adjuvant targeted therapy: A multicentre analysis. Br. J. Cancer 2021, 124, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; McKeown, J.; Dimitriou, F.; Jacques, S.K.; Zimmer, L.; Allayous, C.; Yeoh, H.L.; Haydon, A.; Ressler, J.M.; Galea, C.; et al. Efficacy and safety of “second adjuvant” therapy with BRAF/MEK inhibitors after local therapy for recurrent melanoma following adjuvant PD-1 based immunotherapy. Eur. J. Cancer 2024, 199, 113561. [Google Scholar] [CrossRef]
- Helgadottir, H.; Ni, L.; Ullenhag, G.J.; Felkenius, J.; Mikiver, R.; Bagge, R.O.; Isaksson, K. Survival before and after the introduction of adjuvant treatment in stage III melanoma: A nationwide registry-based study. Ann. Oncol. 2024, 35, S715–S716. [Google Scholar] [CrossRef]
- Ochenduszko, S.; Puskulluoglu, M.; Pacholczak-Madej, R.; Ruiz-Millo, O. Adjuvant anti-PD1 immunotherapy of resected skin melanoma: An example of non-personalized medicine with no overall survival benefit. Crit. Rev. Oncol./Hematol. 2024, 202, 104443. [Google Scholar] [CrossRef]
- Van Akkoi, A.; Mandala, M.; Nathan, P.; Haydon, A.; Postow, M.; Rutkozski, P. Adjuvant systemic therapy in melanoma: Relative versus absolute benefit; the number needed to treat (NNT) versus the number needed to harm (NNH)? EJC Skin Cancer 2024, 2, 10021. [Google Scholar] [CrossRef]
- Ma, E.Z.; Terhune, J.H.; Zafari, Z.; Blackburn, K.W.; Olson, J.A.; Mullins, C.D.; Hu, Y. Treat now or treat later: Comparative effectiveness of adjuvant therapy in resenceted stage IIA melanoma. J. Am. Coll. Surg. 2022, 234, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Stassen, R.C.; Maas, C.; van der Veldt, A.; Lo, S.N.; Saw, R.; Varey, A.; Scolyer, R.; Long, G.V.; Thompson, J.F.; Rutkowski, P.; et al. Development and validation of novel model predict recurrence-free survival and melanoma-specific survival after sentinel lymph node biopsy in retrospective, multicentre analysis. Lancet Oncol. 2024, 25, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Orlowski, R.; Xu, X.; Mick, R.; George, S.; Yan, P.; Manne, S.; Kraye, A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 2019, 25, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Sharon, C.E.; Tortorello, C.N.; Ma, K.L.; Huang, A.S.; Xu, X.; Giles, L.R.; McGettigan, S.; Kreider, K.; Schuchter, L.M.; Mathew, A.J.; et al. Long-term outcomes to neoadjuvant pembrolizumab based on pathological response for patients with resectable stage III/IV cutaneous melanoma. Ann. Oncol. 2023, 34, 806–812. [Google Scholar] [CrossRef]
- Long, G.V.; Saw, R.P.; Lo, S.; Nieweg, O.E.; Shannon, K.F.; Gonzalez, M.; Gumisnki, A.; Lee, J.; Lee, H.; Ferguson, P.M.; et al. Neodajuvant dabrafenib combined with trametinib for resectable, stage IIIB-C, BRAFV600 mutation-positive melanoma (NeoCombi): A single-arm, open-label, single-centre, phase 2 trial. Lancet Oncol. 2019, 20, 961–971. [Google Scholar] [CrossRef]
- Menzies, A.M.; Lo, S.; Saw, R.P.; Gonzalez, M.; Ching, S.; Nieweg, O.E.; Shannon, K.F.; Ferguson, P.M.; Lee, J.; Emmett, L.; et al. Five-year analysis of neoadjuvant dabrafenib and trametinib for stage III melanoma. Ann. Oncol. 2024, 35, 739–746. [Google Scholar] [CrossRef]
- Amaria, R.N.; Prieto, P.A.; Tetzlaff, M.; Reuben, A.; Andrews, M.C.; Ross, M.J.; Gliza, I.C.; Cormier, J.; HJwu, W.J.; Tawbi, H.; et al. Neoadjuvant plus adjuvant dabrafenib and trametinib versus standard of care in patients with high-risk, surgically resectable melanoma: A single-centre, open-label, randomized, phase 2 trial. Lancet Oncol. 2018, 19, 181–193. [Google Scholar] [CrossRef]
- Versluis, J.M.; Menzies, A.M.; Sikorska, K.; Rozeman, E.A.; Saw, R.; van Houdt, W.J.; Eriksson, H.; Klop, W.; Ching, S.; van Thienen, J.V.; et al. Survival update of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanomas in the OpACIN and OpACIN-neo trials. Ann. Oncol. 2023, 34, 420–430. [Google Scholar] [CrossRef]
- Reijers, I.I.; Menzies, A.; van Akkoi, A.; Versluis, J.; van den Heuvel, N.; Saw, R.; Pennington, T.; Kapiteijen, E.; van der Veidt, A.; Suijkerbuijk, K.; et al. Personalized response-directed surgery and adjuvant therapy after neoadjuvant ipilimumab and nivolumab in high-risk stage III melanoma: The PRADO trial. Nat. Med. 2022, 28, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Amaria, R.; Rozeman, E.; Huang, A.C.; Telzlaff, M.T.; van de Wiel, B.; Lo, S.; Tarhini, A.A.; Burton, E.; Pennington, T.E.; et al. Pathological response and survival with neoadjuvant therapy in melanoma: A pooled analysis from the international Neoadjuvant Melanoma Consortium (INMC). Nat. Med. 2021, 27, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Blank, C.U.; Amaria, R.N.; Hieken, T.J.; Sandhu, S.K.; Barros, M.J.; Mitchell, T.C.C.; Eroglu, Z.; Samoylenko, I.V.; Rutkowski, P.; et al. Long-term survival with neoadjuvant therapy in melanoma: Updated pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Ann. Oncol. 2024, 35 (Suppl. S2), LBA41. [Google Scholar] [CrossRef]
- Patel, S.; Olhus, M.; Chen, Y.; Wright, G.P.; Yosl, K.; Hyngstrom, J.R.; Hu-Lieskovan, S.; Lao, C.; Fecher, L.A.; Truong, T.G.; et al. Neoadjuvant-adjuvant or adjuvant-only pembrolizumab in advanced melanoma. N. Engl. J. Med. 2023, 388, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Lucas, M.W.; Scolyer, R.A.; van de Wiel, B.A.; Menzies, A.M.; Lopez-Yurda, M.; Hoeijmakers, L.L.; Saw, R.; Lijnsvelt, J.; Maher, N.G.; et al. Neoadjuvant nivolumab and ipilimumab in resectable stage III melanoma. N. Engl. J. Med. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.W.; Menzies, A.M.; Lopez-Yurda, M.; Scolyer, R.A.; van de Wiel, B.; Saw, R.; van Houdt, W.; Maher, N.; Torres Acosta, A.; Boers-Sonderen, M.; et al. Distantmetastasis-free survival of neoadjuvant nivolumab plus ipilimumab versus adjuvant nivolumab in resectable, macroscopic stage III melanoma. Ann. Oncol. 2024, 35 (Suppl. S2), LBA42. [Google Scholar] [CrossRef]
- Long, G.V.; Robert, C.; Hill, A.G.; Marqueste, C.G.; Portnoy, D.; Shapira, R.; Cohen, J.E.; Khattak, M.E.; Lebbe, C.; Menzies, A.M.; et al. KEYMAKER-U02 substudy 02C: Neoadjuvant pembrolizumab (pembro) and investigational agents followed by adjuvant pembro for stage IIIB-D melanoma. Ann. Oncol. 2024, 35 (Suppl. S2), 10820. [Google Scholar] [CrossRef]
- Slinguff, C.L.; Petroni, G.; Chianese-Bullock, K.; Smolkin, M.E.; Hibbitts, S.; Murphy, C.; Joahnsen, N.; Grosh, W.W.; Yamschichov, G.V.; Neese, P.Y.; et al. Immunologic and clinical outcomes of a randomized phase II trial of two multipeptide vaccines for melanoma in the adjuvant setting. Clin. Cancer Res. 2007, 13, 6386–6395. [Google Scholar] [CrossRef]
- Hu, Y.; Kim, H.; Blackwell, C.; Slingluff, C.L. Long-term outcomes of helper peptide vaccination for metastatic melanoma. Ann. Surg. 2015, 262, 456–464. [Google Scholar] [CrossRef]
- Ninmer, E.K.; Zhu, H.; Chianese, K.B.; van Mehren, M.; Haas, N.B.; Ross, M.I.; Dengel, L.T.; Sling, C.L. Multipeptide vaccines for melanoma in the adjuvant setting: Long-term survival outcomes and post-hoc analysis of a randomized phase II trial. Nat. Commun. 2024, 15, 2570. [Google Scholar] [CrossRef]
- Bal, K.F.; Schreibelt, G.; Bloemendal, M.; van Willigen, W.; Hans-de Bree, S.; de Goede, A.; de Boer, A.; Bos, K.; Dulveman-de Boer, J.; Olde Nordekamp, M.; et al. Adjuvant dendritic cell therapy in stage IIIB/C melanoma: The MIND-DC randomized phase III trial. Nat. Commun. 2024, 15, 1632. [Google Scholar] [CrossRef]
- Weber, J.S.; Carlino, M.S.; Khattak, A.; Meniawy, T.; Ansstas, G.; Taylor, M.H.; Kim, K.B.; McKean, M.; Long, G.V.; Sullivan, R.J.; et al. Individualised neoantigen therapy mRNA-5147 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): A randomized, phase 2b study. Lancet 2024, 403, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Khattak, A.M.; Carlino, M.S.; Meniawy, T.; Taylor, M.H.; Ansstas, G.M.; Kim, K.B.; McKean, M.; Sullivan, R.J.; Faries, M.B.; et al. Individualized neoantigen therapy mRNA-4157 (V940) plus pembrolizumab in resected melanoma: 3-year update from the mRNA-4157-P201 (KEYNOTE-942) trial. J. Clin. Oncol. 2024, 16 (Suppl. S16), LBA9512. [Google Scholar] [CrossRef]
- Weber, J.S.; Luke, J.J.; Khattak, A.M.; Carlino, M.S.; Meehan, R.S.; Brown, M.; Zhang, J.; Krepler, C.; Duic, J.P.; Long, G.V.; et al. INTerpath-001: Pembrolizumab with V940 (mRAN-4157) versus pembrolizumab with placebo for adjuvant treatment of high-risk stage II-IV melanoma. J. Clin. Oncol. 2024, 42 (Suppl. S16), TPS9616. [Google Scholar] [CrossRef]
- Gainor, J.F.; Patel, M.R.; Weber, J.S.; Gutierrez, M.; Bauman, J.E.; Clarke, J.M.; Julian, R.; Scott, A.J.; Geiger, J.L.; Kirtane, K.; et al. T-cell responses to individualized neoantigen therapy mRNA-4157 (V940) alone or in combination with pembrolizumab in the phase 1 KEYNOTE-603 study. Cancer Discov. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Rutkowski, P.O.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Queirolo, P.; Dummer, R.; Butler, M.O.; Hill, A.G.; et al. Final, 10-year outcomes with Nivolumab plus Ipilimumab in advanced melanoma. N. Engl. J. Med. 2024, in press. [Google Scholar] [CrossRef]
- Varaljai, R.; Zimmer, L.; Al-Matary, Y.; Kaptein, P.; Albrecht, L.J.; Shannan, B.; Brase, J.C.; Gusenleitner, D.; Amaral, T.; Wyss, N.; et al. Interleukin 17 signaling supports clinical benefit of dual CTLA-4 and PD-1 checkpoint inhibition in melanoma. Nat. Cancer 2023, 4, 1292–1308. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Forsyth, P.A.; Algazi, A.; Hamid, O.; Hodi, F.S.; Moschos, S.J.; Khushalani, N.I.; Lewis, K.; Lao, C.D.; Postow, M.A.; et al. Combined Nivolumab and Ipilimumab in melanoma mestastatic to the brain. N. Engl. J. Med. 2018, 379, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.; Forsyth, P.; Algazi, A.; Hamid, O.; Lao, C.D.; Mochos, S.J.; Atkins, M.B.; Lewis, K.; Postow, M.A.; Thomas, R.P.; et al. Long-term outcomes of patients with active melanoma brain metastases treated with combination nivolumab plus ipilimumab (CheckMate 204): Final results of an open-label, multicentre, phase 2 study. Lancet Oncol. 2021, 22, 1692–1704. [Google Scholar] [CrossRef]
- Kattenhøj, K.D.; Møberg, C.L.; Guldbrandt, L.M.; Friis, R.B.; Mapendano, C.K.; Petersen, S.K.; Ruhlmann, C.H.B.; Svane, I.M.; Donia, M.; Ellebaek, E.; et al. Efficacy of Ipilimumab and Nivolumab in patients with melanoma and brain metastases—A Danish real-world cohort. Cancers 2024, 16, 2559. [Google Scholar] [CrossRef]
- Di Giacomo, A.M.; Chiarion-Sileni, V.; del Vecchio, M.; Ferrucci, P.F.; Guida, M.; Quaglino, P.; Guidoboni, M.; Marchetti, P.; Simonetti, E.; Santangelo, F.; et al. Nivolumab plus ipilimumab in melanoma patients with asymptomatic brain metastases: 7-year outcomes and quality of life from the multicenter phase III NIBIT-M2 trial. Eur. J. Cancer 2024, 199, 113531. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Casula, M.; Bulgarelli, J.; Pisano, M.; Piccinini, C.; Piccin, L.; Cossu, A.; Mandalà, M.; Ferrucci, P.F.; Guidoboni, M.; et al. Sequential immunotherapy and targeted therapy for metastatic BRAF V600 mutated melanoma: 4-year survival and biomarkers evaluation from the phase III SECOMBIT trial. Nat. Commun. 2024, 15, 146. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Mandalà, M.; Ferrucci, P.F.; Guidoboni, M.; Rutkowski, P.; Ferraresi, V.; Arance, A.; Guida, M.; Maiello, E.; Gogas, H.; et al. Sequencing of checkpoint or BRAF/MEK inhibitors on brain metastases in melanoma. NEJM Evid. 2014, 3, EVIUDoa2400087. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination dabrafenib and trametinib versus combination nivolumab and ipilimumab for patients with advanced BRAF-mutant melanoma: The DREAMseq trial-ECOG-ACRIN EA6134. J. Clin. Oncol. 2023, 41, 186–197. [Google Scholar] [CrossRef]
- Dummer, R.; Queirolo, P.; Duhard, P.G.; Hu, Y.; Wang, D.; de Azevedo, S.J.; Robert, C.; Ascierto, P.A.; Chiarion-Sileni, V.; Pronzato, P.; et al. Atezolizumab, vemurafenib, and cobimetinib in patients with melanoma with CNS metastases (TRICOTEL): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2023, 24, e461–e471. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo-Gutierrez, E.; Rutkopwski, P.; Gogas, H.J.; Lao, C.D.; de Menezes, J.J.; et al. Relatimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 2022, 386, e461–e471. [Google Scholar] [CrossRef]
- Long, G.V.; Hodi, S.; Lipson, E.J.; Schadendorf, D.; Ascierto, P.A.; Matamala, L.; Salman, P.; Castillo-Gutierrez, E.; Rutkowski, P.; Gogas, H.J.; et al. Overall survival and response with nivolumab and relatlimab in advanced melanoma. NEJM Evid. 2023, 2, EVIDoa2200239. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Hodi, S.; Schadendorf, D.; Ascierto, P.A.; Mammala, L.; Castillo-Gutierrez, E.; Rutkowski, P.; Gogas, H.; Lao, C.D.; Menez, J.; et al. Nivolumab (NIVO) plus relatlimab (RELA) in previously untreated metastatic or unresectable melanoma (RELATIVITY-047): Overall survival (OS) and melanoma specific survival (MSS) outcomes at 3 years. J. Clin. Oncol. 2024, 42 (Suppl. S16), 9524. [Google Scholar] [CrossRef]
- Dolfi, S.; Tang, T.; Long, G.; Ascierto, P.; Hodi, S.; Lipson, E.; Schadendorf, D.; Wojcik, J.; Postelnek, J.; Wang, Y.; et al. Biomarker analyses of baseline tumor specimens and on-treatment changes in sera samples of patients enrolled in the RELATIVITY-047 trial to characterize LAG-3 biology. J. Immunother. Cancer 2023, 10 (Suppl. S2), 606. [Google Scholar]
- Lipson, E.J.; Dolfi, S.; Tang, H.; Gogas, H.; Tawbi, H.A.; Hodi, F.S.; Ascierto, P.A.; Gutierrez, E.C.; Schadendorf, D.; Medina Soto, F.A.; et al. Unraveling relatlimab (RELA)-specific biology using biomarker analyses in patients with advanced melanoma treated with nivolumab (NIVO)+RELA or NIVO alone in RELATIVITY-047. Ann. Oncol. 2023, 34 (Suppl. S2), LBAS1. [Google Scholar] [CrossRef]
- Long, G.V.; Lipson, E.J.; Hodi, S.; Ascierto, P.A.; Larkin, J.; Lao, C.; Grob, J.J.; Ejzykowicz, F.; Moshyk, A.; Garcia-Horton, V.; et al. First-line nivolumab plus relatlimab versus nivolumab plus ipilimumab in advanced melanoma: An indirect treatment comparison using RELATIVITY-047 and CheckMate 067 trial data. J. Clin. Oncol. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Lipson, E.J.; Dummer, R.; Larkin, J.; Long, G.V.; Sanborn, R.E.; Chiarion-Sileni, V.; Dreno, B.; Dalle, S.; Schadendorf, D.; et al. Nivolumab and relatlimab in patients with advanced melanoma that had progressed on anti-programmed death-1/programmed death ligand 1 therapy: Results from the phase I/IIa RELATIVITY-020 trial. J. Clin. Oncol. 2023, 41, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Dummer, R.; Gaudy-Marqueste, C.; Bowyer, S.; Lipson, E.J.; Ghisoni, E.; Middleton, M.R.; Ratto, B.; Jackson, W.J.; Cheong, A.; et al. Efficacy and safety of triplet nivolumab, relatlimab and ipilimumab (NIVO+RELA+IPI) in advanced melanoma: Results from RELATIVITY-048. J. Clin. Oncol. 2024, 42 (Suppl. S16), 9504. [Google Scholar] [CrossRef]
- Dummer, R.; Long, G.V.; Robert, C.; Tawby, H.; Flaherty, K.T.; Ascierto, P.A.; Nathan, P.D.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; et al. Randomized phase III trial evaluating spartalizumab plus dabrafenib and trametinib for BRAFV600-mutant unersectable or metastatic melanoma. J. Clin. Oncol. 2022, 40, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Flaherty, K.T.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; et al. Update on tolerability and overall survival in COLUMBUS: Landmark analysis of a randomized phase 3 trial of encorafenib plus binimetinib vs. vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. Eur. J. Cancer 2020, 126, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Groot, J.W.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsova, I.; Liszkay, G.; et al. COLUMBUS 5-year update: A randomized, open-label, phase III trial of encorafenib pluis binimetinib versus vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. J. Clin. Oncol. 2022, 40, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Grroot, J.W.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Karjsova, I.; et al. COLUMBUS 7-year update: A randomized, open-label, phase III trial of encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF V600E/K-mutant melanoma. EUR J. Cancer 2024, 204, 114073. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R. Contribution of MEK inhibition to BRAF/MEK inhibitor combination treatment of BRAF-mutant melanoma: Part 2 of the randomized, open-label phase III COLUMBUS trial. J. Clin. Oncol. 2023, 41, 4621–4631. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Dutriaux, C.; Oppong, F.; Kicinski, M.; Routier, E.; Neidhardt, E.M.; Durand, X.; Barooudjian, B.; Saiag, P.; Gaudy-Marqueste, C.; et al. Combination of encorafenib and binemitinib followed by ipilimumab and nivolumab versus ipilimumab and nivolumab in patients with advanced BRAF-V600E/K-mutated melanoma: The primary analysis of an EORTC randomized phase II study (EBIN). J. Clin. Oncol. 2024, 42 (Suppl. S16), LBA9503. [Google Scholar] [CrossRef]
- Samaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a tumorinfiltrating lymphocyte therapy, in metastatic melanoma. J. Clin. Oncol. 2021, 39, 2656–2666. [Google Scholar]
- Medina, T.; Chesney, J.A.; Whitman, E.; Kluger, H.; Thomas, S.; Sarnaik, A.A.; Kirkwood, J.M.; Larkin, J.; Weber, J.; Hamid, O.; et al. Long-term efficacy and safety of lifileucel tumor-infiltrating lymphocyte (TIL) cell tehrapy in patients with advanced melanoma: A 4-year analysis of the C-144-01 study. J. Immunother. Cancer 2023, 11 (Suppl. S1), A1–A1731. [Google Scholar]
- Rohaan, M.W.; Borch, T.H.; van den Bergh, G.H.; Met, O.; Kessels, R.; Geukes Foppen, M.H.; Granhoj, J.S.; Nuijen, B.; Mijenhuis, C.; Jedema, I.; et al. Tumor-infiltrating lymphocyte therapy or ipilimumab in advanced lymphoma. N. Engl. J. Med. 2022, 387, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.S.; Gogas, H.; Hong, Y.K.; In, G.K.; de Speville Uribe, B.D.; Furness, A.; Castano, A.G.; Haeflinger, S.; He, K.; Medina, T.; et al. Efficacy and safety of Lifileucel, an autologous tumor-infiltrating lymphocyte cell therapy, and pembrolizumab in patients with immune checkpoint inhibitor-naïve unresectable or metastatic melanoma: Updated results from IOV-COM-202 cohort 1A. J. Clin. Oncol. 2024, 42 (Suppl. S16), 9505. [Google Scholar] [CrossRef]
- Martin-Lluesma, S.; Svane, I.M.; Dafni, U.; Vervita, K.; Karlis, D.; Dimopoulou, G.; Tsourti, Z.; Rohaan, M.W.; Haanen, J.B.; Coukos, G. Efficacy of TIL therapy in advanced cutaneous melanoma in the current immune-oncology era: Updated systematic review and meta-analysis. Ann. Oncol. 2024, 35, 860–872. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Clinical Trial | Patients | Treatment | RFS | DMFS | Safety | Parameters Correlating with Response |
|---|---|---|---|---|---|---|
| KEYNOTE-716 Phase III double-blind, randomized | 976 stage IIB/C 487 (Pembro) 489 (Placebo) | Adjuvant pembroluzumab (PE) vs. placebo (PL) | 36 months All 76.2%(PE) 63.4%(PL) IIB 79.7%(PE) 66.5% (PL) IIC 72.4%(PE) 58% PL) | 36 months All 84.4%(PE) 74.7%(PL) IIB 86.7%(PE) 78.9%(PL) IIC 80.9%(PE) 68.1%(PL) | Grade 3–4 17.2%(PE) 5.1% (PL) | Not reported |
| CHECK MATE 76k Phase III double-blind, randomized | 790 stage IIB/C 526 (Nivo) 264 (Placebo) | Adjuvant nivolumab (NI) vs. placebo (PL) | 12 months All 89%(NI) 79% (PL) BRAF-WT 91.2% (NI) 77.1% (PL) BRAF-mut 87.3% (NI) 81.7% (PL) | 12 months All 92.3%(NI) 86.7%(PL) IIC 87.9%(NI) 78.7%(PL) | Grade 3–4 10.3% (NI) 2.3% (PL) | Higher IFN-γ signature and % CD8+ cells |
| Clinical Study | Patients | Treatment | RFS | DMFS | OS | Rate of Recurrence |
|---|---|---|---|---|---|---|
| Bai et al. Multicenter, retrospective cohort study | 598 stage III BRAF-mutant melanoma | 393 pts dabrafenib plus trametinib (DT) 205 pts anti-PD1 (PD1) | At 33 months DT 51 months PD1 44.8 months | Not reported | At 3 years DT 74.4% PD1 77.9% | Progression DT 45% PD1 27.7% Distant metastases DT 20% PD1 26% |
| Bloem et al. Dutch melanoma treatment registry nation-wide cohort | 416 Two groups of 213 propensity score-matched stage IIIB BRAF-mutant patients | 213 pts DT 213 pts anti-PD1 | At 2 years DT 66.1% PD1 70.2% | At 2 years DT 84.1% PD1 82.1% | At 2 years DT80.4% PD1 85.1% | DT 34% PD1 30% |
| Clinical Trial | Patients | Treatment | PFS | OS | Safety | Parameters Correlating with Response |
|---|---|---|---|---|---|---|
| CHECK MATE 067 NCT 01844505, phase III, randomized | 945 metastatic 314 (Nivo+Ipi) 316 (Nivo) 315 (ipi) | Nivo+Ipi Nivo Ipi Follow-up 10 years | Nivo+Ipi 11.5 mo Nivo 6.9 mo Ipi 2.9 mo | OS at 10 yr Nivo+Ipi 71.9 mo Nivo 36.9 mo Ipi 19.9 mo MSS Nivo+Ipi >120 mo Nivo 49.4 mo Ipi 21.9 mo | Grade 3–4 Nivo+Ipi 59% Nivo 23% Ipi 29% | BRAF-mut respond to Nivo+Ipi better than BRAF-WT; response to Nivo-Ipi is associated with TH17 signatures |
| CHECK MATE 204 NCT 02320058, phase II, open label, multicenter | 119 with brain metastases 101 asymptomatic (cohort A) 18 symptomatic (cohort B) | Nivo+Ipi 36 months follow-up | 36 mo intracranial Cohort A 57.4% Cohort B 18.9% | Cohort A 71.9% Cohort B 36.6% | Grade 3–4 5% | Not reported |
| SECOMBIT NCT 02631447, phase II, randomized | 206 patients with BRAFV600 metastatic Arm A (69) Arm B (69) Arm C (68) | Arm A Enco+Bini→Nivo+Ipi Arm B Nivo+Ipi→Enco+Bini Arm C Enco+Bini 8wk; Nivo+Ipi→Enco+Bini | At 4 years Arm A 29% Arm B 55% Arm C 54% | At 4 years Arm A 46% Arm B 64% Arm C 59% | Not reported | Improved OS in patients with JAK mutations and low IFN-γ serum levels |
| RELATIVITY-047 NCT 03470922, phase II-III double blind, randomized | 714 metastatic Nivo+Rela (355) Nivo (359) | Nivo+Rela Nivo | At 5 years Nivo+Rela 48.7% Nivo 39.4% | At 5 years Nivo+Rel 27.7% Nivo 21.6% | Grade 3–4 Nivo+Ipi 22% Nivo 12% | Improved response to Nivo+Rela in high baseline PD1+CD8+ and ICOS1+CD8+ T cells |
| Clinical Trial | Patients | Treatment | PFS | OS | Safety | Parameters Correlating with Response |
|---|---|---|---|---|---|---|
| C-144-01 NCT 02360579, nonrandomized, phase II | 153 advanced melanoma, ICI refractory | Lifileucel (autologous TIL) >1 × 109 cells | ORR 31.4% | mOS 13.9 months 1 yr 54% 2 yr 33.9% 3 yr 28.3% 4 yr 22.2% | Grade 3–4 100% | Few responses in patients with high TMB and brain and liver metastases |
| M14 TIL NCT 092278887 | 168 advanced melanoma (86% ICI refractory) 84 TILs 84 ipilimumab (Ipi) | Autologous TILs At least 5 × 109 cells | TIL 7.2 months Ipi 3.1 months | mOS TIL 25.8 months Ipi 18.9 months | Grade 3–4 TIL 100% Ipi 57% | Not Reported |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelosi, E.; Castelli, G.; Testa, U. Braf-Mutant Melanomas: Biology and Therapy. Curr. Oncol. 2024, 31, 7711-7737. https://doi.org/10.3390/curroncol31120568
Pelosi E, Castelli G, Testa U. Braf-Mutant Melanomas: Biology and Therapy. Current Oncology. 2024; 31(12):7711-7737. https://doi.org/10.3390/curroncol31120568
Chicago/Turabian StylePelosi, Elvira, Germana Castelli, and Ugo Testa. 2024. "Braf-Mutant Melanomas: Biology and Therapy" Current Oncology 31, no. 12: 7711-7737. https://doi.org/10.3390/curroncol31120568
APA StylePelosi, E., Castelli, G., & Testa, U. (2024). Braf-Mutant Melanomas: Biology and Therapy. Current Oncology, 31(12), 7711-7737. https://doi.org/10.3390/curroncol31120568