IDH Mutant Cholangiocarcinoma: Pathogenesis, Management, and Future Therapies


Abstract
:1. Introduction
2. Isocitrate Dehydrogenase
3. Pathogenesis of IDH Mutant Cholangiocarcinoma
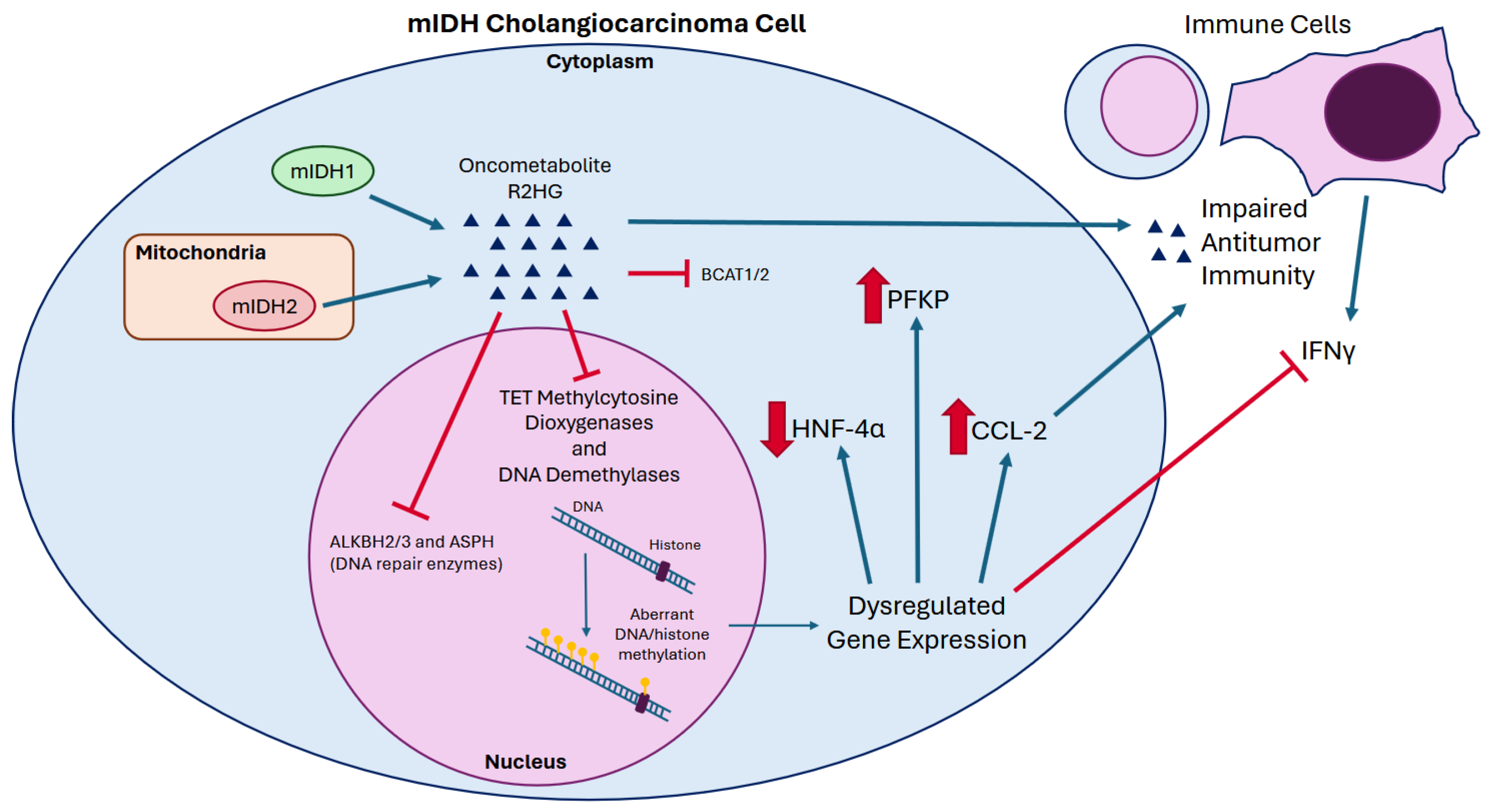
3.1. Mutant IDH and Oncometabolite R2HG
3.2. R2HG Drives Epigenetic Dysregulation and Oncogenesis
3.3. R2HG and the Immune Microenvironment
4. Clinical Characteristics of mIDH Cholangiocarcinoma
5. mIDH Directed Therapies
5.1. Ivosidenib
5.2. Olutasidenib
5.3. IDH305
5.4. TQB3454
5.5. LY3410738
5.6. HMPL-306
6. Other Novel Therapeutic Strategies
6.1. mIDH Inhibitors and Cytotoxic Chemotherapy
6.2. mIDH Inhibitors and Immune Checkpoint Inhibitor Therapy
6.3. Src Kinase Inhibition
6.4. PARP Inhibition
6.5. Chloroquine and Metformin
6.6. Other Agents
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Javle, M.; Lee, S.; Azad, N.S.; Borad, M.J.; Kate Kelley, R.; Sivaraman, S.; Teschemaker, A.; Chopra, I.; Janjan, N.; Parasuraman, S.; et al. Temporal Changes in Cholangiocarcinoma Incidence and Mortality in the United States from 2001 to 2017. Oncologist 2022, 27, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Storandt, M.H.; Tella, S.H.; Wieczorek, M.A.; Hodge, D.; Elrod, J.K.; Rosenberg, P.S.; Jin, Z.; Mahipal, A. Projected Incidence of Hepatobiliary Cancers and Trends Based on Age, Race, and Gender in the United States. Cancers 2024, 16, 684. [Google Scholar] [CrossRef]
- Valle, J.W.; Kelley, R.K.; Nervi, B.; Oh, D.-Y.; Zhu, A.X. Biliary Tract Cancer. Lancet 2021, 397, 428–444. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network Biliary Tract Cancers (Version 4.2024). Available online: https://www.nccn.org/professionals/physician_gls/pdf/btc.pdf (accessed on 30 September 2024).
- Oh, D.-Y.; Ruth He, A.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Ah Lee, M.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Ueno, M.; Yoo, C.; Finn, R.S.; Furuse, J.; Ren, Z.; Yau, T.; Klümpen, H.-J.; Chan, S.L.; Ozaka, M.; et al. Pembrolizumab in Combination with Gemcitabine and Cisplatin Compared with Gemcitabine and Cisplatin Alone for Patients with Advanced Biliary Tract Cancer (KEYNOTE-966): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 401, 1853–1865. [Google Scholar] [CrossRef]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Second-Line FOLFOX Chemotherapy versus Active Symptom Control for Advanced Biliary Tract Cancer (ABC-06): A Phase 3, Open-Label, Randomised, Controlled Trial. Lancet Oncol. 2021, 22, 690–701. [Google Scholar] [CrossRef]
- Verdaguer, H.; Saurí, T.; Acosta, D.A.; Guardiola, M.; Sierra, A.; Hernando, J.; Nuciforo, P.; Miquel, J.M.; Molero, C.; Peiró, S.; et al. ESMO Scale for Clinical Actionability of Molecular Targets Driving Targeted Treatment in Patients with Cholangiocarcinoma. Clin. Cancer Res. 2022, 28, 1662–1671. [Google Scholar] [CrossRef]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Sinha, C.; Vats, P.; Tran, N.; Robinson, D.R.; Gunchick, V.; Wu, Y.-M.; Cao, X.; Ning, Y.; Wang, R.; Rabban, E.; et al. Genomics Driven Precision Oncology in Advanced Biliary Tract Cancer Improves Survival. Neoplasia 2023, 42, 100910. [Google Scholar] [CrossRef] [PubMed]
- Carosi, F.; Broseghini, E.; Fabbri, L.; Corradi, G.; Gili, R.; Forte, V.; Roncarati, R.; Filippini, D.M.; Ferracin, M. Targeting Isocitrate Dehydrogenase (IDH) in Solid Tumors: Current Evidence and Future Perspectives. Cancers 2024, 16, 2752. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Yan, H. Isocitrate Dehydrogenase 1 and 2 Mutations in Cancer: Alterations at a Crossroads of Cellular Metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef]
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-Associated Mutation and beyond: The Emerging Biology of Isocitrate Dehydrogenases in Human Disease. Sci. Adv. 2019, 5, eaaw4543. [Google Scholar] [CrossRef]
- Chen, X.; Ding, J. Molecular Insights into the Catalysis and Regulation of Mammalian NAD-Dependent Isocitrate Dehydrogenases. Curr. Opin. Struct. Biol. 2023, 82, 102672. [Google Scholar] [CrossRef]
- Cloos, P.A.C.; Christensen, J.; Agger, K.; Helin, K. Erasing the Methyl Mark: Histone Demethylases at the Center of Cellular Differentiation and Disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Ruiz, B.I.; Lowman, X.H.; Yang, Y.; Fan, Q.; Wang, T.; Wu, H.; Hanse, E.A.; Kong, M. Alpha-Ketoglutarate Regulates Tnfrsf12a/Fn14 Expression via Histone Modification and Prevents Cancer-Induced Cachexia. Genes 2023, 14, 1818. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-Ketoglutarate Maintains the Pluripotency of Embryonic Stem Cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef]
- Tran, T.Q.; Hanse, E.A.; Habowski, A.N.; Li, H.; Gabra, M.B.I.; Yang, Y.; Lowman, X.H.; Ooi, A.M.; Liao, S.Y.; Edwards, R.A.; et al. α-Ketoglutarate Attenuates Wnt Signaling and Drives Differentiation in Colorectal Cancer. Nat. Cancer 2020, 1, 345–358. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-Derived Mutations in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1α. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Herr, C.Q.; Hausinger, R.P. Amazing Diversity in Biochemical Roles of Fe(II)/2-Oxoglutarate Oxygenases. Trends Biochem. Sci. 2018, 43, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Koh, H.-J.; Park, D.-C.; Song, B.J.; Huh, T.-L.; Park, J.-W. Cytosolic NADP+-Dependent Isocitrate Dehydrogenase Status Modulates Oxidative Damage to Cells. Free. Radic. Biol. Med. 2002, 32, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.-H.; Son, M.-K.; Koh, H.-J.; Lee, S.-M.; Song, I.-H.; Kim, Y.-O.; Lee, Y.-S.; Jeong, K.-S.; Kim, W.B.; Park, J.-W.; et al. Control of Mitochondrial Redox Balance and Cellular Defense against Oxidative Damage by Mitochondrial NADP+-Dependent Isocitrate Dehydrogenase. J. Biol. Chem. 2001, 276, 16168–16176. [Google Scholar] [CrossRef]
- Koh, H.-J.; Lee, S.-M.; Son, B.-G.; Lee, S.-H.; Ryoo, Z.Y.; Chang, K.-T.; Park, J.-W.; Park, D.-C.; Song, B.J.; Veech, R.L.; et al. Cytosolic NADP+-Dependent Isocitrate Dehydrogenase Plays a Key Role in Lipid Metabolism. J. Biol. Chem. 2004, 279, 39968–39974. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, P.A.; Laviolette, L.A.; Kelleher, J.K.; Iliopoulos, O.; Stephanopoulos, G. Cofactor Balance by Nicotinamide Nucleotide Transhydrogenase (NNT) Coordinates Reductive Carboxylation and Glucose Catabolism in the Tricarboxylic Acid (TCA) Cycle. J. Biol. Chem. 2013, 288, 12967–12977. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Gu, Y.; Zhang, F.; Zhao, Y.; Yuan, Y.; Hao, Z.; Sheng, Y.; Li, W.Y.; Wakeham, A.; Cairns, R.A.; et al. IDH1 Deficiency Attenuates Gluconeogenesis in Mouse Liver by Impairing Amino Acid Utilization. Proc. Natl. Acad. Sci. USA 2017, 114, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Itsumi, M.; Inoue, S.; Elia, A.J.; Murakami, K.; Sasaki, M.; Lind, E.F.; Brenner, D.; Harris, I.S.; Chio, I.I.C.; Afzal, S.; et al. Idh1 Protects Murine Hepatocytes from Endotoxin-Induced Oxidative Stress by Regulating the Intracellular NADP+/NADPH Ratio. Cell Death Differ. 2015, 22, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.J.; Ahn, Y.; Lee, J.H.; Park, K.M.; Park, J.-W. IDH2 Deficiency Promotes Mitochondrial Dysfunction and Cardiac Hypertrophy in Mice. Free Radic. Biol. Med. 2015, 80, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Parachoniak, C.A.; Bardeesy, N. IDH Mutations in Liver Cell Plasticity and Biliary Cancer. Cell Cycle 2014, 13, 3176–3182. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Rendina, A.R.; Pietrak, B.; Smallwood, A.; Zhao, H.; Qi, H.; Quinn, C.; Adams, N.D.; Concha, N.; Duraiswami, C.; Thrall, S.H.; et al. Mutant IDH1 Enhances the Production of 2-Hydroxyglutarate Due to Its Kinetic Mechanism. Biochemistry 2013, 52, 4563–4577. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.-L.; Mashimo, T.; Raisanen, J.M.; Marin-Valencia, I.; et al. 2-Hydroxyglutarate Detection by Magnetic Resonance Spectroscopy in IDH-Mutated Patients with Gliomas. Nat. Med. 2012, 18, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-Associated Metabolite 2-Hydroxyglutarate Accumulates in Acute Myelogenous Leukemia with Isocitrate Dehydrogenase 1 and 2 Mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of Antitumor T Cell Immunity by the Oncometabolite (R)-2-Hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Goyal, L.; Yau, T.; Poon, R.T.; Ancukiewicz, M.; Deshpande, V.; Christiani, D.C.; Liebman, H.M.; Yang, H.; Kim, H.; et al. Circulating Oncometabolite 2-Hydroxyglutarate Is a Potential Surrogate Biomarker in Patients with Isocitrate Dehydrogenase-Mutant Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2014, 20, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.L.; O’Kane, G.M.; Mason, W.P.; Zhang, W.-J.; Spiliopoulou, P.; Hansen, A.R.; Grant, R.C.; Knox, J.J.; Stockley, T.L.; Zadeh, G.; et al. Circulating Oncometabolite 2-Hydroxyglutarate as a Potential Biomarker for Isocitrate Dehydrogenase (IDH1/2) Mutant Cholangiocarcinoma. Mol. Cancer Ther. 2024, 23, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Abou-Alfa, G.K.; Zhu, A.X.; Pandya, S.S.; Jia, H.; Yin, F.; Gliser, C.; Hua, Z.; Hossain, M.; Yang, H. Pharmacokinetics/Pharmacodynamics of Ivosidenib in Advanced IDH1-Mutant Cholangiocarcinoma: Findings from the Phase III ClarIDHy Study. Cancer Chemother. Pharmacol. 2024, 93, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Mellinghoff, I.K.; Wen, P.Y.; Lowery, M.A.; Goyal, L.; Tap, W.D.; Pandya, S.S.; Manyak, E.; Jiang, L.; Liu, G.; et al. Clinical Pharmacokinetics and Pharmacodynamics of Ivosidenib, an Oral, Targeted Inhibitor of Mutant IDH1, in Patients with Advanced Solid Tumors. Investig. New Drugs 2020, 38, 433–444. [Google Scholar] [CrossRef]
- Brunner, A.M.; Neuberg, D.S.; Wander, S.A.; Sadrzadeh, H.; Ballen, K.K.; Amrein, P.C.; Attar, E.; Hobbs, G.S.; Chen, Y.-B.; Perry, A.; et al. Isocitrate Dehydrogenase 1 and 2 Mutations, 2-Hydroxyglutarate Levels, and Response to Standard Chemotherapy for Patients with Newly Diagnosed Acute Myeloid Leukemia. Cancer 2019, 125, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH Mutation Impairs Histone Demethylation and Results in a Block to Cell Differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.-M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The Oncometabolite 2-Hydroxyglutarate Inhibits Histone Lysine Demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef] [PubMed]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Burgess, K.; Bhargava, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-Oncogenic Role of Mutant IDH2 in Leukemia Initiation and Maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Steinhäuser, S.; Silva, P.; Lenk, L.; Beder, T.; Hartmann, A.; Hänzelmann, S.; Fransecky, L.; Neumann, M.; Bastian, L.; Lipinski, S.; et al. Isocitrate Dehydrogenase 1 Mutation Drives Leukemogenesis by PDGFRA Activation Due to Insulator Disruption in Acute Myeloid Leukemia (AML). Leukemia 2023, 37, 134–142. [Google Scholar] [CrossRef]
- Rahme, G.J.; Javed, N.M.; Puorro, K.L.; Xin, S.; Hovestadt, V.; Johnstone, S.E.; Bernstein, B.E. Modeling Epigenetic Lesions That Cause Gliomas. Cell 2023, 186, 3674–3685.e14. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) Mutation Increases Murine Haematopoietic Progenitors and Alters Epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 Promotes Leukemogenesis in Vivo and Can Be Specifically Targeted in Human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Tanaka, S.; Curry, W.T.; Loebel, F.; Zhao, D.; Tateishi, K.; Chen, J.; Klofas, L.K.; Lelic, N.; Kim, J.C.; et al. Targetable Signaling Pathway Mutations Are Associated with Malignant Phenotype in IDH-Mutant Gliomas. Clin. Cancer Res. 2014, 20, 2898–2909. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, Y.; Katsumoto, T.; Aikawa, Y.; Shima, Y.; Kagiyama, Y.; Soga, T.; Matsunaga, H.; Seki, T.; Araki, K.; Kitabayashi, I. IDH2 and NPM1 Mutations Cooperate to Activate Hoxa9/Meis1 and Hypoxia Pathways in Acute Myeloid Leukemia. Cancer Res. 2015, 75, 2005–2016. [Google Scholar] [CrossRef]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH Inhibits HNF-4α to Block Hepatocyte Differentiation and Promote Biliary Cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.-F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.-X.; et al. Mutations in Isocitrate Dehydrogenase 1 and 2 Occur Frequently in Intrahepatic Cholangiocarcinomas and Share Hypermethylation Targets with Glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef]
- Goeppert, B.; Toth, R.; Singer, S.; Albrecht, T.; Lipka, D.B.; Lutsik, P.; Brocks, D.; Baehr, M.; Muecke, O.; Assenov, Y.; et al. Integrative Analysis Defines Distinct Prognostic Subgroups of Intrahepatic Cholangiocarcinoma. Hepatology 2019, 69, 2091–2106. [Google Scholar] [CrossRef]
- Liao, H.; Chen, X.; Wang, H.; Lin, Y.; Chen, L.; Yuan, K.; Liao, M.; Jiang, H.; Peng, J.; Wu, Z.; et al. Whole-Genome DNA Methylation Profiling of Intrahepatic Cholangiocarcinoma Reveals Prognostic Subtypes with Distinct Biological Drivers. Cancer Res. 2024, 84, 1747–1763. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Lü, L.; Lin, P.; Chen, Z.; Quan, Z.; Tang, Z. Multiple Cellular Origins and Molecular Evolution of Intrahepatic Cholangiocarcinoma. Cancer Lett. 2016, 379, 253–261. [Google Scholar] [CrossRef]
- Fujiwara, H.; Tateishi, K.; Misumi, K.; Hayashi, A.; Igarashi, K.; Kato, H.; Nakatsuka, T.; Suzuki, N.; Yamamoto, K.; Kudo, Y.; et al. Mutant IDH1 Confers Resistance to Energy Stress in Normal Biliary Cells through PFKP-Induced Aerobic Glycolysis and AMPK Activation. Sci. Rep. 2019, 9, 18859. [Google Scholar] [CrossRef]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.-L.; Ye, D.; et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef]
- Nagaoka, K.; Bai, X.; Liu, D.; Cao, K.; Mulla, J.; Ji, C.; Chen, H.; Nisar, M.A.; Bay, A.; Mueller, W.; et al. Elevated 2-Oxoglutarate Antagonizes DNA Damage Responses in Cholangiocarcinoma Chemotherapy through Regulating Aspartate Beta-Hydroxylase. Cancer Lett. 2024, 580, 216493. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Cho, Y.; Kim, J.H.; Kim, J.; Nam, H.Y.; Kim, S.W.; Son, J. Branched-Chain Amino Acids Sustain Pancreatic Cancer Growth by Regulating Lipid Metabolism. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, A.; Osawa, T.; Noda, M.; Kobayashi, Y.; Aki, S.; Nakano, Y.; Saito, T.; Shimizu, D.; Komatsu, H.; Sugaya, M.; et al. Convergent Genomic Diversity and Novel BCAA Metabolism in Intrahepatic Cholangiocarcinoma. Br. J. Cancer 2023, 128, 2206–2217. [Google Scholar] [CrossRef]
- Rimini, M.; Fabregat-Franco, C.; Burgio, V.; Lonardi, S.; Niger, M.; Scartozzi, M.; Rapposelli, I.G.; Aprile, G.; Ratti, F.; Pedica, F.; et al. Molecular Profile and Its Clinical Impact of IDH1 Mutated versus IDH1 Wild Type Intrahepatic Cholangiocarcinoma. Sci. Rep. 2022, 12, 18775. [Google Scholar] [CrossRef]
- Wu, M.-J.; Shi, L.; Merritt, J.; Zhu, A.X.; Bardeesy, N. Biology of IDH Mutant Cholangiocarcinoma. Hepatology 2022, 75, 1322–1337. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Cao, L.; Chen, R.; Ye, C.; Li, Q.; Jiang, Q.; Yan, F.; Wan, M.; Zhang, X.; Ruan, J. Differential Isocitrate Dehydrogenase 1 and Isocitrate Dehydrogenase 2 Mutation-Related Landscape in Intrahepatic Cholangiocarcinoma. Oncologist 2024, 29, e1061–e1072. [Google Scholar] [CrossRef]
- Makawita, S.; Lee, S.; Kong, E.; Kwong, L.N.; Abouelfetouh, Z.; Danner De Armas, A.; Xiao, L.; Murugesan, K.; Danziger, N.; Pavlick, D.; et al. Comprehensive Immunogenomic Profiling of IDH1-/2-Altered Cholangiocarcinoma. JCO Precis. Oncol. 2024, 8, e2300544. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Robertson, S.A.; Hong, S.-M.; Felsenstein, M.; Anders, R.A.; Pea, A.; Nottegar, A.; Veronese, N.; He, J.; Weiss, M.J.; et al. PBRM1 Loss Is a Late Event during the Development of Cholangiocarcinoma. Histopathology 2017, 71, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Hirosawa, T.; Ishida, M.; Ishii, K.; Kanehara, K.; Kudo, K.; Ohnuma, S.; Kamei, T.; Motoi, F.; Naitoh, T.; Selaru, F.M.; et al. Loss of BAP1 Expression Is Associated with Genetic Mutation and Can Predict Outcomes in Gallbladder Cancer. PLoS ONE 2018, 13, e0206643. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Friedland, S.C.; Alexander, W.; Myers, J.A.; Wang, W.; O’Dell, M.R.; Getman, M.; Whitney-Miller, C.L.; Agostini-Vulaj, D.; Huber, A.R.; et al. Arid1a Mutation Suppresses TGF-β Signaling and Induces Cholangiocarcinoma. Cell Rep. 2022, 40, 111253. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Wu, M.-J.; Shi, L.; Dubrot, J.; Merritt, J.; Vijay, V.; Wei, T.-Y.; Kessler, E.; Olander, K.E.; Adil, R.; Pankaj, A.; et al. Mutant IDH Inhibits IFNγ-TET2 Signaling to Promote Immunoevasion and Tumor Maintenance in Cholangiocarcinoma. Cancer Discov. 2022, 12, 812–835. [Google Scholar] [CrossRef] [PubMed]
- Zabransky, D.J.; Kartalia, E.; Lee, J.W.; Leatherman, J.M.; Charmsaz, S.; Young, S.E.; Chhabra, Y.; Franch-Expósito, S.; Kang, M.; Maru, S.; et al. Tumor-Derived CCL2 Drives Tumor Growth and Immunosuppression in IDH1-Mutant Cholangiocarcinoma. Hepatology 2024. [Google Scholar] [CrossRef] [PubMed]
- Job, S.; Rapoud, D.; Dos Santos, A.; Gonzalez, P.; Desterke, C.; Pascal, G.; Elarouci, N.; Ayadi, M.; Adam, R.; Azoulay, D.; et al. Identification of Four Immune Subtypes Characterized by Distinct Composition and Functions of Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 72, 965–981. [Google Scholar] [CrossRef]
- Xiang, X.; Liu, Z.; Zhang, C.; Li, Z.; Gao, J.; Zhang, C.; Cao, Q.; Cheng, J.; Liu, H.; Chen, D.; et al. IDH Mutation Subgroup Status Associates with Intratumor Heterogeneity and the Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Adv. Sci. 2021, 8, 2101230. [Google Scholar] [CrossRef] [PubMed]
- Aguado-Fraile, E.; Tassinari, A.; Ishii, Y.; Sigel, C.; Lowery, M.A.; Goyal, L.; Gliser, C.; Jiang, L.; Pandya, S.S.; Wu, B.; et al. Molecular and Morphological Changes Induced by Ivosidenib Correlate with Efficacy in Mutant-IDH1 Cholangiocarcinoma. Future Oncol. 2021, 17, 2057–2074. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-J.; Kondo, H.; Kammula, A.V.; Shi, L.; Xiao, Y.; Dhiab, S.; Xu, Q.; Slater, C.J.; Avila, O.I.; Merritt, J.; et al. Mutant IDH1 Inhibition Induces dsDNA Sensing to Activate Tumor Immunity. Science 2024, 385, eadl6173. [Google Scholar] [CrossRef] [PubMed]
- Kipp, B.R.; Voss, J.S.; Kerr, S.E.; Barr Fritcher, E.G.; Graham, R.P.; Zhang, L.; Highsmith, W.E.; Zhang, J.; Roberts, L.R.; Gores, G.J.; et al. Isocitrate Dehydrogenase 1 and 2 Mutations in Cholangiocarcinoma. Hum. Pathol. 2012, 43, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified Through Broad-Based Tumor Genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef]
- Krell, D.; Assoku, M.; Galloway, M.; Mulholland, P.; Tomlinson, I.; Bardella, C. Screen for IDH1, IDH2, IDH3, D2HGDH and L2HGDH Mutations in Glioblastoma. PLoS ONE 2011, 6, e19868. [Google Scholar] [CrossRef] [PubMed]
- Boscoe, A.N.; Rolland, C.; Kelley, R.K. Frequency and Prognostic Significance of Isocitrate Dehydrogenase 1 Mutations in Cholangiocarcinoma: A Systematic Literature Review. J. Gastrointest. Oncol. 2019, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.; Hai Nguyen, H.; et al. Whole-Genome Mutational Landscape of Liver Cancers Displaying Biliary Phenotype Reveals Hepatitis Impact and Molecular Diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef] [PubMed]
- Chan-on, W.; Nairismägi, M.-L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome Sequencing Identifies Distinct Mutational Patterns in Liver Fluke–Related and Non-Infection-Related Bile Duct Cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, Histomorphological and Molecular Classification of Cholangiocarcinoma. Liver Int. 2019, 39, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Drill, E.; Vakiani, E.; Pak, L.M.; Boerner, T.; Askan, G.; Schvartzman, J.M.; Simpson, A.L.; Jarnagin, W.R.; Sigel, C.S. Distinct Histomorphological Features Are Associated with IDH1 Mutation in Intrahepatic Cholangiocarcinoma. Hum. Pathol. 2019, 91, 19–25. [Google Scholar] [CrossRef]
- Ma, B.; Meng, H.; Tian, Y.; Wang, Y.; Song, T.; Zhang, T.; Wu, Q.; Cui, Y.; Li, H.; Zhang, W.; et al. Distinct Clinical and Prognostic Implication of IDH1/2 Mutation and Other Most Frequent Mutations in Large Duct and Small Duct Subtypes of Intrahepatic Cholangiocarcinoma. BMC Cancer 2020, 20, 318. [Google Scholar] [CrossRef]
- Lee, K.; Song, Y.S.; Shin, Y.; Wen, X.; Kim, Y.; Cho, N.-Y.; Bae, J.M.; Kang, G.H. Intrahepatic Cholangiocarcinomas with IDH1/2 Mutation-Associated Hypermethylation at Selective Genes and Their Clinicopathological Features. Sci. Rep. 2020, 10, 15820. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Z.; Ding, Z.; Dong, W.; Liang, H.; Chu, L.; Zhang, B.; Chen, X. IDH1 Mutation Correlates with a Beneficial Prognosis and Suppresses Tumor Growth in IHCC. J. Surg. Res. 2018, 231, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Govindan, A.; Sheth, R.A.; Nardi, V.; Blaszkowsky, L.S.; Faris, J.E.; Clark, J.W.; Ryan, D.P.; Kwak, E.L.; Allen, J.N.; et al. Prognosis and Clinicopathologic Features of Patients with Advanced Stage Isocitrate Dehydrogenase (IDH) Mutant and IDH Wild-Type Intrahepatic Cholangiocarcinoma. Oncologist 2015, 20, 1019–1027. [Google Scholar] [CrossRef]
- Kinzler, M.N.; Jeroch, J.; Klasen, C.; Himmelsbach, V.; Koch, C.; Finkelmeier, F.; Trojan, J.; Zeuzem, S.; Pession, U.; Reis, H.; et al. Impact of IDH1 Mutation on Clinical Course of Patients with Intrahepatic Cholangiocarcinoma: A Retrospective Analysis from a German Tertiary Center. J. Cancer Res. Clin. Oncol. 2023, 149, 6391–6398. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network Central Nervous System Cancers (Version 3.2024). Available online: https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf (accessed on 18 December 2024).
- Niger, M.; Nichetti, F.; Casadei-Gardini, A.; Rizzato, M.D.; Pircher, C.; Bini, M.; Franza, A.; Rimini, M.; Burgio, V.; Sposetti, C.; et al. Platinum Sensitivity in Patients with IDH1/2 Mutated vs Wild-Type Intrahepatic Cholangiocarcinoma: A Propensity Score-Based Study. Int. J. Cancer 2022, 151, 1310–1320. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Burris, H.A.; Janku, F.; Shroff, R.T.; Cleary, J.M.; Azad, N.S.; Goyal, L.; Maher, E.A.; Gore, L.; Hollebecque, A.; et al. Safety and Activity of Ivosidenib in Patients with IDH1-Mutant Advanced Cholangiocarcinoma: A Phase 1 Study. Lancet Gastroenterol. Hepatol. 2019, 4, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-Mutant, Chemotherapy-Refractory Cholangiocarcinoma (ClarIDHy): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients with Advanced Cholangiocarcinoma with IDH1 Mutation. JAMA Oncol. 2021, 7, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Casak, S.J.; Pradhan, S.; Fashoyin-Aje, L.A.; Ren, Y.; Shen, Y.-L.; Xu, Y.; Chow, E.C.Y.; Xiong, Y.; Zirklelbach, J.F.; Liu, J.; et al. FDA Approval Summary: Ivosidenib for the Treatment of Patients with Advanced Unresectable or Metastatic, Chemotherapy Refractory Cholangiocarcinoma with an IDH1 Mutation. Clin. Cancer Res. 2022, 28, 2733–2737. [Google Scholar] [CrossRef]
- Rimini, M.; Burgio, V.; Antonuzzo, L.; Rimassa, L.; Oneda, E.; Soldà, C.; Cito, P.; Nasti, G.; Lavacchi, D.; Zanuso, V.; et al. Updated Survival Outcomes with Ivosidenib in Patients with Previously Treated IDH1-Mutated Intrahepatic-Cholangiocarcinoma: An Italian Real-World Experience. Ther. Adv. Med. Oncol. 2023, 15, 17588359231171574. [Google Scholar] [CrossRef]
- Choe, S.; Wang, H.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular Mechanisms Mediating Relapse Following Ivosidenib Monotherapy in IDH1-Mutant Relapsed or Refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef]
- Wang, F.; Morita, K.; DiNardo, C.D.; Furudate, K.; Tanaka, T.; Yan, Y.; Patel, K.P.; MacBeth, K.J.; Wu, B.; Liu, G.; et al. Leukemia Stemness and Co-Occurring Mutations Drive Resistance to IDH Inhibitors in Acute Myeloid Leukemia. Nat. Commun. 2021, 12, 2607. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schvartzman, J.M.; Hou, S.; Famulare, C.; Patel, M.; Roshal, M.; Do, R.K.; Zehir, A.; et al. Isoform Switching as a Mechanism of Acquired Resistance to Mutant Isocitrate Dehydrogenase Inhibition. Cancer Discov. 2018, 8, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Rouaisnel, B.; Daina, A.; Raghavan, S.; Roller, L.A.; Huffman, B.M.; Singh, H.; Wen, P.Y.; Bardeesy, N.; Zoete, V.; et al. Secondary IDH1 Resistance Mutations and Oncogenic IDH2 Mutations Cause Acquired Resistance to Ivosidenib in Cholangiocarcinoma. NPJ Precis. Oncol. 2022, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.-M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-Based Design and Identification of FT-2102 (Olutasidenib), a Potent Mutant-Selective IDH1 Inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.; Watts, J. Olutasidenib: From Bench to Bedside. Blood Adv. 2023, 7, 4358–4365. [Google Scholar] [CrossRef]
- Jones, R.L.; Macarulla, T.; Charlson, J.A.; Van Tine, B.A.; Goyal, L.; Italiano, A.; Massard, C.; Rosenthal, M.; De La Fuente, M.I.; Roxburgh, P.; et al. A Phase Ib/II Study of Olutasidenib in Patients with Relapsed/Refractory IDH1 Mutant Solid Tumors: Safety and Efficacy as Single Agent. J. Clin. Oncol. 2020, 38, e16643. [Google Scholar] [CrossRef]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and Evaluation of Clinical Candidate IDH305, a Brain Penetrant Mutant IDH1 Inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Schimmer, A.D.; Yee, K.W.L.; Hochhaus, A.; Kraemer, A.; Carvajal, R.D.; Janku, F.; Bedard, P.; Carpio, C.; Wick, A.; et al. A Phase I Study of IDH305 in Patients with Advanced Malignancies Including Relapsed/Refractory AML and MDS That Harbor IDH1R132 Mutations. Blood 2016, 128, 1073. [Google Scholar] [CrossRef]
- Yuan, J.; Zheng, Y.; Xu, L.; Xie, F.; Gu, S.; Li, Q.; Zhang, J.; Ba, Y.; Huaxin, D.; Yang, A.; et al. 286P TQB3454 in Mutant IDH1 Advanced Cholangiocarcinoma (CCA): Results of a Phase I Dose Escalation and Expansion Study Cohort. Ann. Oncol. 2024, 35, S120. [Google Scholar] [CrossRef]
- Luk, I.S.; Bridgwater, C.M.; Yu, A.; Boila, L.D.; Yáñez-Bartolomé, M.; Lampano, A.E.; Hulahan, T.S.; Boukhali, M.; Kathiresan, M.; Macarulla, T.; et al. SRC Inhibition Enables Formation of a Growth Suppressive MAGI1-PP2A Complex in Isocitrate Dehydrogenase-Mutant Cholangiocarcinoma. Sci. Transl. Med. 2024, 16, eadj7685. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M.; Pilat, M.J.; Uboha, N.; Azad, N.S.; Cho, M.; Davis, E.J.; Ahnert, J.R.; Tinoco, G.; Shapiro, G.I.; Khagi, S.; et al. Abstract B040: NCI 10129: A Phase 2 Study of the PARP Inhibitor Olaparib (AZD2281) in IDH1 and IDH2 Mutant Advanced Solid Tumors. Mol. Cancer Ther. 2023, 22, B040. [Google Scholar] [CrossRef]
- Tsang, E.S.; O’Kane, G.M.; Knox, J.J.; Chen, E.X. A Phase II Study of Olaparib and Durvalumab in Patients with IDH-Mutated Cholangiocarcinoma. J. Clin. Oncol. 2023, 41, 4099. [Google Scholar] [CrossRef]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 Mutations Alter Citric Acid Cycle Metabolism and Increase Dependence on Oxidative Mitochondrial Metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Hochhaus, A.; Frattini, M.G.; Yee, K.; Zander, T.; Krämer, A.; Chen, X.; Ji, Y.; Parikh, N.S.; Choi, J.; et al. A Phase 1 Study of IDH305 in Patients with IDH1R132-Mutant Acute Myeloid Leukemia or Myelodysplastic Syndrome. J. Cancer Res. Clin. Oncol. 2023, 149, 1145–1158. [Google Scholar] [CrossRef]
- Harding, J.; Ikeda, M.; Goyal, L.; Rodon, J.; Bai, L.; Oh, D.; Park, J.; Chen, L.; Ueno, M.; Liao, C.; et al. SO-1 A First-in-Human Phase 1 Study of LY3410738, a Covalent Inhibitor of Mutant IDH1 and IDH2, as Monotherapy and in Combination with Cisplatin and Gemcitabine in Advanced IDH-Mutant Cholangiocarcinoma. Ann. Oncol. 2023, 34, S161. [Google Scholar] [CrossRef]
- Janku, F.; Kauh, J.S.; Tucci, C.; Yang, Z.; Kania, M.K.; Alese, O.B. A Multicenter Open-Label Phase 1 Study Evaluating the Safety and Tolerability of HMPL-306 in Patients with Locally Advanced or Metastatic Solid Tumors with IDH Mutations. J. Clin. Oncol. 2021, 39, TPS3159. [Google Scholar] [CrossRef]
- Yang, N.; Hu, J.; Li, T.; Yu, J.; Shi, D.; Cheng, M.; Zhong, Z.; Wang, J.; Sai, Y.; Qing, W.; et al. Abstract 543: Preclinical Characteristic of HMPL-306, a CNS-Penetrable Dual Inhibitor of Mutant IDH1 and IDH2. Cancer Res. 2023, 83, 543. [Google Scholar] [CrossRef]
- Dong, L.-Q.; Shi, Y.; Ma, L.-J.; Yang, L.-X.; Wang, X.-Y.; Zhang, S.; Wang, Z.-C.; Duan, M.; Zhang, Z.; Liu, L.-Z.; et al. Spatial and Temporal Clonal Evolution of Intrahepatic Cholangiocarcinoma. J. Hepatol. 2018, 69, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Rimini, M.; Loi, E.; Fabregat-Franco, C.; Burgio, V.; Lonardi, S.; Niger, M.; Scartozzi, M.; Raposelli, I.G.; Aprile, G.; Ratti, F.; et al. Next-Generation Sequencing Analysis of Cholangiocarcinoma Identifies Distinct IDH1-Mutated Clusters. Eur. J. Cancer 2022, 175, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Khurshed, M.; Aarnoudse, N.; Hulsbos, R.; Hira, V.V.V.; van Laarhoven, H.W.M.; Wilmink, J.W.; Molenaar, R.J.; van Noorden, C.J.F. IDH1-Mutant Cancer Cells Are Sensitive to Cisplatin and an IDH1-Mutant Inhibitor Counteracts This Sensitivity. FASEB J. 2018, 32, 6344–6352. [Google Scholar] [CrossRef]
- Lachowiez, C.; DiNardo, C.D.; Stein, E. Combining Isocitrate Dehydrogenase Inhibitors with Existing Regimens in Acute Myeloid Leukemia: An Evolving Treatment Landscape. Cancer J. 2022, 28, 21. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Cleary, J.M.; Sahai, V.; Baretti, M.; Bridgewater, J.A.; Hua, Z.; Gliser, C.; Bian, Y.; Abou-Alfa, G.K. A Phase 1/2, Safety Lead-in and Dose Expansion, Open-Label, Multicenter Trial Investigating the Safety, Tolerability, and Preliminary Activity of Ivosidenib in Combination with Nivolumab and Ipilimumab in Previously Treated Subjects with IDH1-Mutated Nonresectable or Metastatic Cholangiocarcinoma. J. Clin. Oncol. 2024, 42, TPS4197. [Google Scholar] [CrossRef]
- Saha, S.K.; Gordan, J.D.; Kleinstiver, B.P.; Vu, P.; Najem, M.S.; Yeo, J.-C.; Shi, L.; Kato, Y.; Levin, R.S.; Webber, J.T.; et al. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC-Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2016, 6, 727–739. [Google Scholar] [CrossRef]
- Mohan, A.; Quingalahua, E.; Gunchick, V.; Paul, S.; Kumar-Sinha, C.; Crysler, O.; Zalupski, M.M.; Sahai, V. PARP Inhibitor Therapy in Patients with IDH1 Mutated Cholangiocarcinoma. Oncologist 2024, 29, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Fanucci, K.; Pilat, M.J.; Shyr, D.; Shyr, Y.; Boerner, S.; Li, J.; Durecki, D.; Drappatz, J.; Puduvalli, V.; Lieberman, F.S.; et al. Multicenter Phase II Trial of the PARP Inhibitor Olaparib in Recurrent IDH1- and IDH2-Mutant Glioma. Cancer Res. Commun. 2023, 3, 192. [Google Scholar] [CrossRef]
- Salamanca-Cardona, L.; Shah, H.; Poot, A.J.; Correa, F.M.; Di Gialleonardo, V.; Lui, H.; Miloushev, V.Z.; Granlund, K.L.; Tee, S.S.; Cross, J.R.; et al. In Vivo Imaging of Glutamine Metabolism to the Oncometabolite 2-Hydroxyglutarate in IDH1/2 Mutant Tumors. Cell Metab. 2017, 26, 830–841.e3. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Corominas-Faja, B.; Rodríguez-Gallego, E.; Bosch-Barrera, J.; Martin-Castillo, B.; De Llorens, R.; Joven, J.; Menendez, J.A. Oncometabolic Mutation IDH1 R132H Confers a Metformin-Hypersensitive Phenotype. Oncotarget 2015, 6, 12279–12296. [Google Scholar] [CrossRef] [PubMed]
- Jarzyna, R.; Lenarcik, E.; Bryła, J. Chloroquine Is a Potent Inhibitor of Glutamate Dehydrogenase in Liver and Kidney-Cortex of Rabbit. Pharmacol. Res. 1997, 35, 79–84. [Google Scholar] [CrossRef]
- Khurshed, M.; Molenaar, R.J.; van Linde, M.E.; Mathôt, R.A.; Struys, E.A.; van Wezel, T.; van Noorden, C.J.F.; Klümpen, H.-J.; Bovée, J.V.M.G.; Wilmink, J.W. A Phase Ib Clinical Trial of Metformin and Chloroquine in Patients with IDH1-Mutated Solid Tumors. Cancers 2021, 13, 2474. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, A.S.; Stein, E.M.; Fathi, A.T.; Frankfurt, O.; Schuh, A.C.; Döhner, H.; Martinelli, G.; Patel, P.A.; Raffoux, E.; et al. Mutant Isocitrate Dehydrogenase 1 Inhibitor Ivosidenib in Combination with Azacitidine for Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2021, 39, 57–65. [Google Scholar] [CrossRef]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycka, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.-P.; et al. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2022, 386, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xiang, H.; Zhang, W. Review of Various NAMPT Inhibitors for the Treatment of Cancer. Front. Pharmacol. 2022, 13, 970553. [Google Scholar] [CrossRef]
- Pang, Y.; Li, Q.; Sergi, Z.; Yu, G.; Sang, X.; Kim, O.; Wang, H.; Ranjan, A.; Merchant, M.; Oudit, B.; et al. Exploiting the Therapeutic Vulnerability of IDH-Mutant Gliomas with Zotiraciclib. bioRxiv 2024. [Google Scholar] [CrossRef]
- Chen, K.-A.; Huang, W.-M.; Chen, E.Y.-T.; Ho, P.-K.; Chueh, C.-H.; Wen, Y.-W.; Chen, M.-H.; Chiang, N.-J.; Tsai, Y.-W. Cost-Effectiveness of Ivosidenib versus Chemotherapy for Previously Treated IDH1-Mutant Advanced Intrahepatic Cholangiocarcinoma in Taiwan. BMC Cancer 2024, 24, 622. [Google Scholar] [CrossRef]
- 3 Committee Discussion|Ivosidenib for Treating Advanced Cholangiocarcinoma with an IDH1 R132 Mutation After 1 or More Systemic Treatments|Guidance|NICE. Available online: https://www.nice.org.uk/guidance/ta948/chapter/3-Committee-discussion#cost-effectiveness-estimates (accessed on 8 January 2025).
- Gervaso, L.; Pellicori, S.; Fazio, N. Ivosidenib for Advanced IDH1-Mutant Cholangiocarcinoma. Lancet Oncol. 2020, 21, e370. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Agent | Trial | Phase (n *) | Median Prior Lines of Therapy (Range) | ORR (%) | DCR (%) | Other Efficacy Endpoints | Citation |
|---|---|---|---|---|---|---|---|
| mIDH1 Inhibitors | |||||||
| Ivosidenib | NCT02073994 | Phase I (73) | 2 (1–5) | 4 (5) | 45 (62) | Median PFS 3.8 mo Median OS 13.8 mo | [106] |
| Ivosidenib | NCT02989857 (ClarIDHy) | Phase III (124) | 1 (1–2) | 3 (2) | 66 (53) | Median PFS 2.7 mo Median OS 10.3 mo | [107,108] |
| Olutasidenib | NCT03684811 | Phase Ib/II (26) | 2 (1–10) | 0 (0) | 6 (23) | NR | [109] |
| IDH305 | NCT02381886 | Phase I (24) | NR | NR | NR | NR | [110] |
| TQB3454 | NCT04481607 | Phase I (33) | ≥1 (range NR ‡) | 3 (9.1) | 22 (66.7) | Median PFS 4.7 mo Median OS 16.1 mo | [111] |
| Dual mIDH1/2 Inhibitors | |||||||
| LY3410738 | NCT04521686 | Phase I (42) | 2 (1–7) | 2 (4) | 23 (56) | Median PFS 3.5 mo | [112] |
| HMPL-306 | NCT04762602 | Phase 1 (ongoing) | NR | NR | NR | NR | [113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bray, A.; Sahai, V. IDH Mutant Cholangiocarcinoma: Pathogenesis, Management, and Future Therapies. Curr. Oncol. 2025, 32, 44. https://doi.org/10.3390/curroncol32010044
Bray A, Sahai V. IDH Mutant Cholangiocarcinoma: Pathogenesis, Management, and Future Therapies. Current Oncology. 2025; 32(1):44. https://doi.org/10.3390/curroncol32010044
Chicago/Turabian StyleBray, Alexander, and Vaibhav Sahai. 2025. "IDH Mutant Cholangiocarcinoma: Pathogenesis, Management, and Future Therapies" Current Oncology 32, no. 1: 44. https://doi.org/10.3390/curroncol32010044
APA StyleBray, A., & Sahai, V. (2025). IDH Mutant Cholangiocarcinoma: Pathogenesis, Management, and Future Therapies. Current Oncology, 32(1), 44. https://doi.org/10.3390/curroncol32010044