Tumor-Associated Extracellular Matrix Obstacles for CAR-T Cell Therapy: Approaches to Overcoming

 ,
,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. ECM as Major Component of Tumor Stroma: Composition/Structure, Properties, and Contribution to Pathogenesis of Cancer
2.1. Composition, Structure, and Properties of Intratumoral ECM
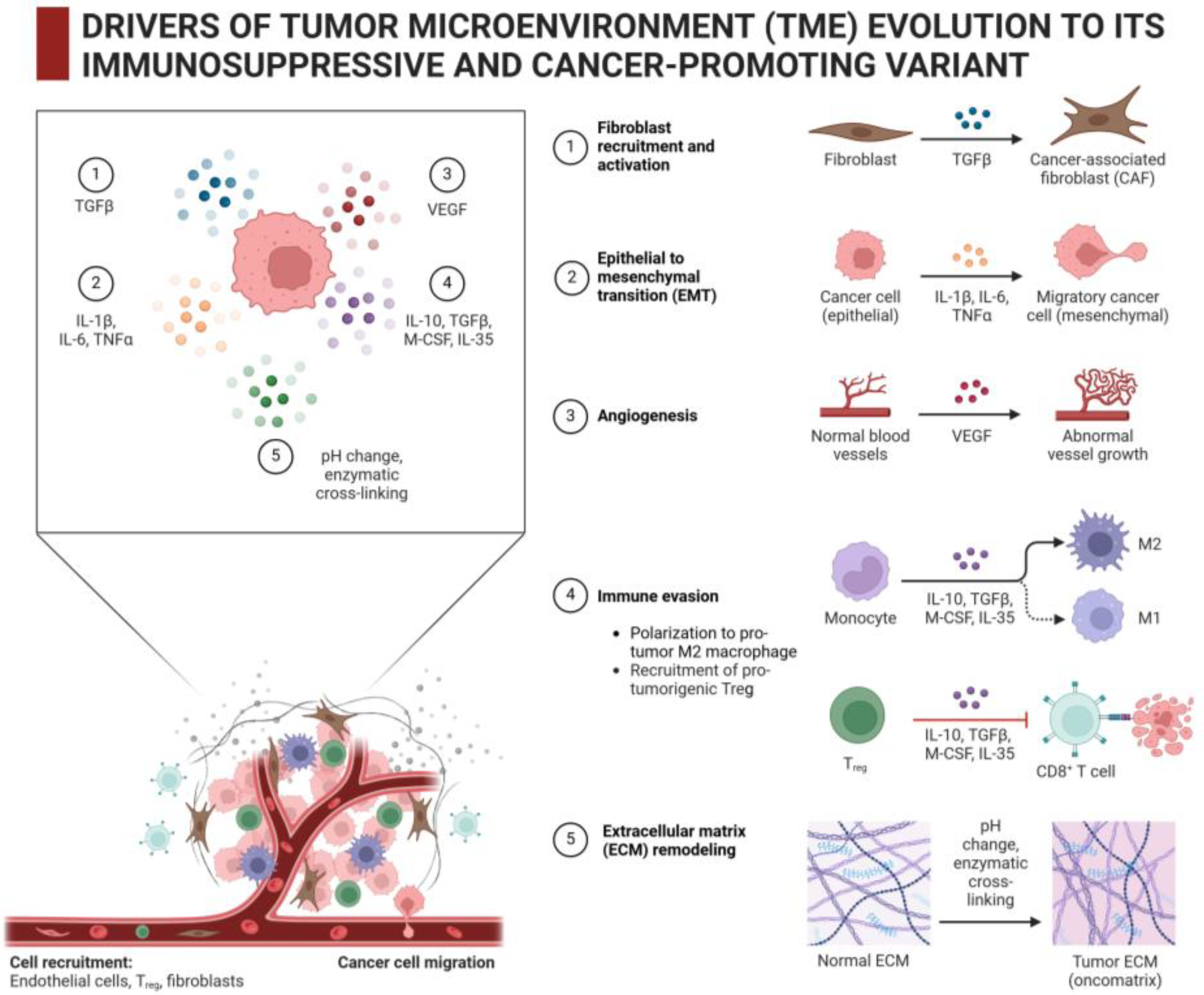
2.2. Which Cells Produce and Modify Intratumoral ECM and What Are Their Stimuli/Drivers?
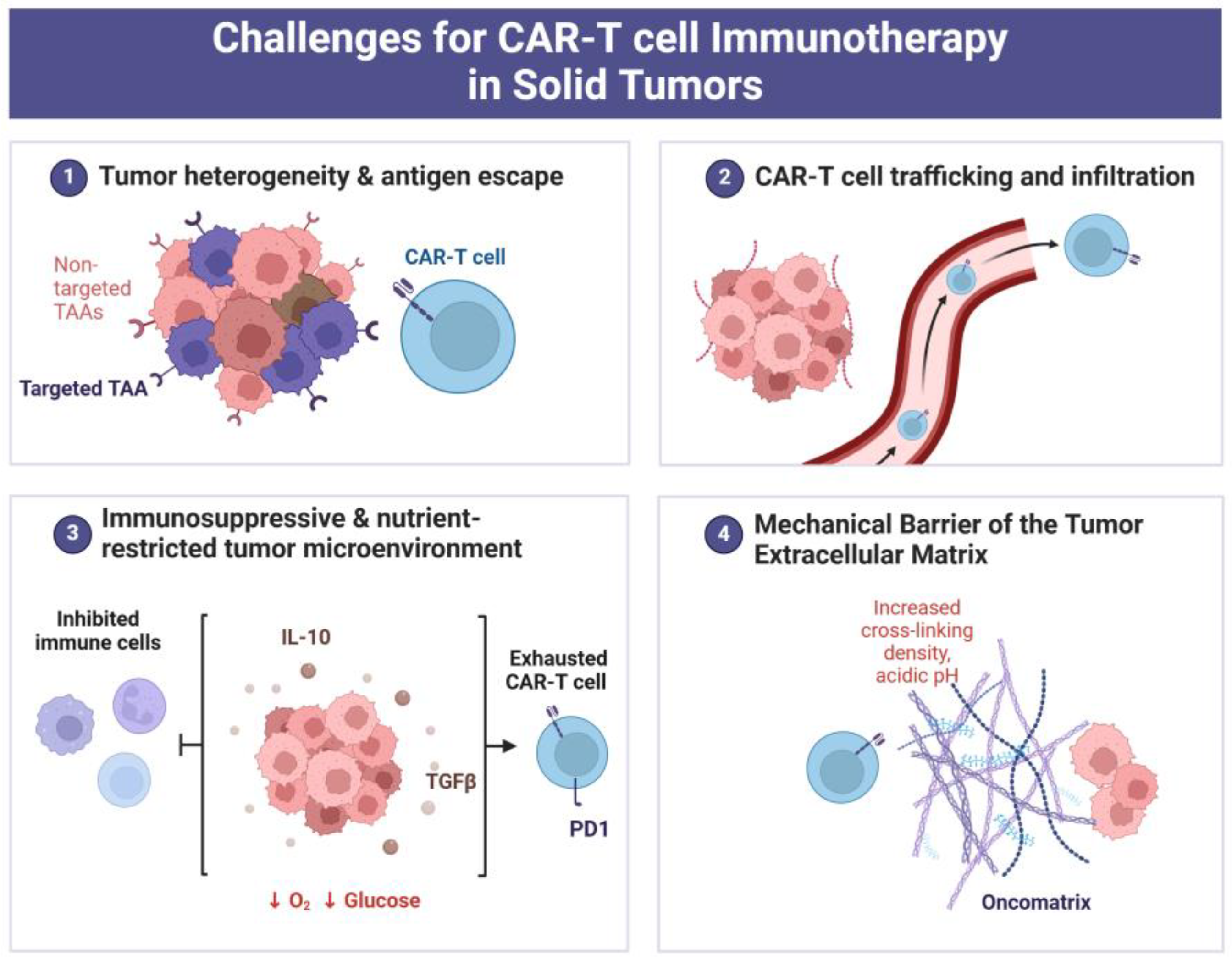
3. Intratumoral ECM as an Obstacle for CAR-T Therapy
3.1. Immunosuppressive Properties of Intratumoral ECM That Can Cause CAR-T Cell Exhaustion/Dysfunction
3.2. ECM Stiffness as Factor That Interferes with T-Lymphocyte Infiltration into Tumor
4. How to Overcome ECM-Associated Barriers in CAR-T Therapy?
4.1. Ex Vivo Preconditioning or Engineering T-Lymphocytes for Their Better Infiltration into Solid Tumors
4.1.1. Preconditioning and Inhibitory Treatments of T-Lymphocytes
4.1.2. Use of Artificial Hydrogels
4.1.3. Gene Engineering Tumor-Directed CAR-T Cells for Their Better Infiltration into Solid Tumors
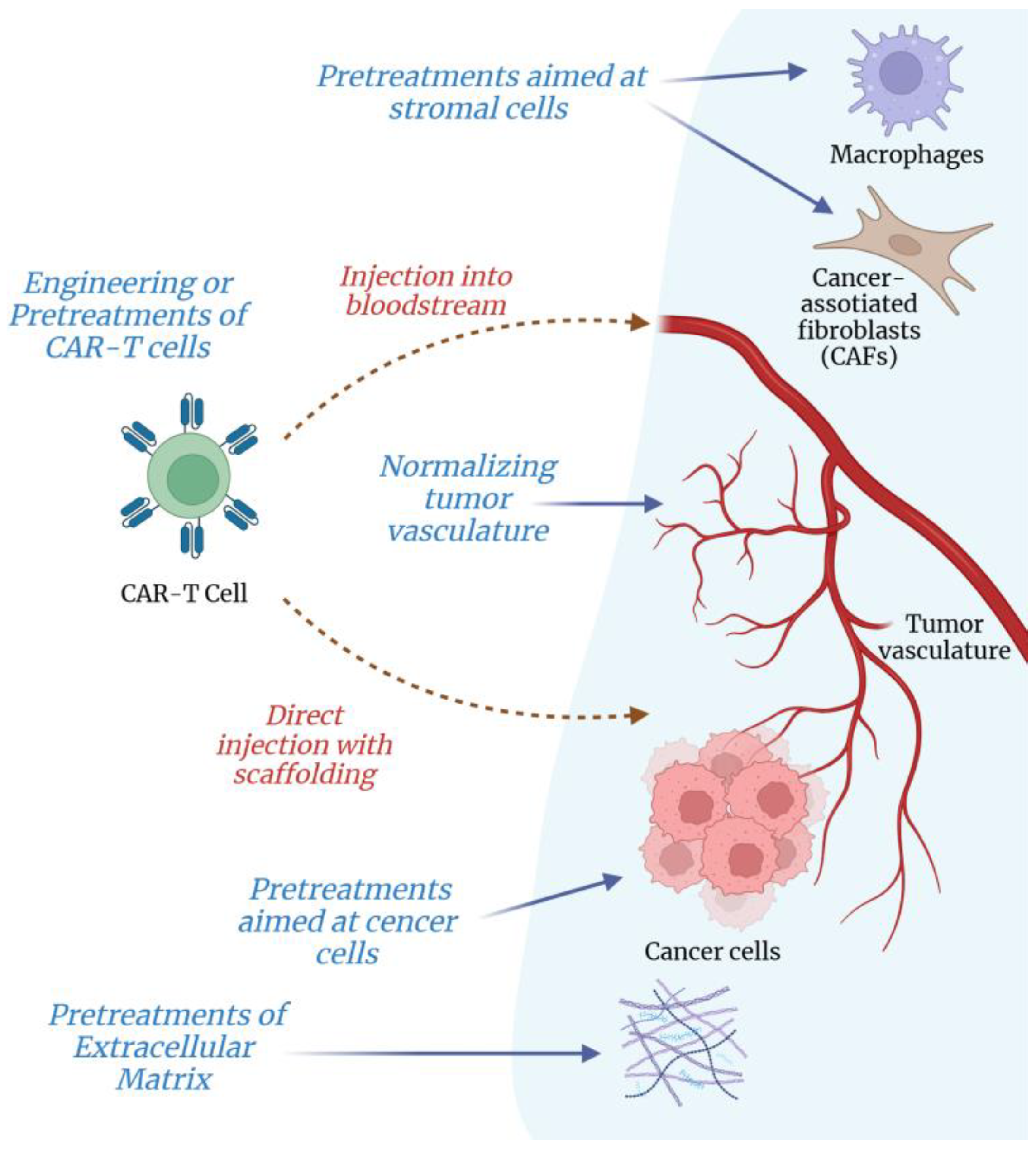
4.2. Treatments of Intratumoral ECM to Improve T-Lymphocyte Infiltration and Killing of Cancer Cells
4.2.1. Pretreatments of Intratumoral ECM
4.2.2. Targeting Cancer and Stromal Cells to Attenuate ECM Synthesis and Fibrosis in Tumor
4.2.3. Normalizing Tumor Vasculature to Make Its Subendothelial Basement Membrane More Penetrable for T-Lymphocytes and CAR-T Cells
4.2.4. Other Potential Agents to Target Tumor-Associated ECM: Oncolytic Viruses, Biotechnological and Nano-Technological Devices … What Else?
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AHR | Aryl hydrocarbon receptor |
| CAF | Cancer-associated fibroblast |
| CAR | Chimeric antigen receptor |
| CSC | Cancer stem cell |
| ECM | Extracellular matrix |
| EMT | Epithelial-to-mesenchymal transition |
| EGF | Epidermal growth factor |
| FAK | Focal adhesion kinase |
| FGF | Fibroblast growth factor |
| GAG | Glycosaminoglycan |
| HDAC | Histone deacetylase |
| HIF | Hypoxia-inducible factor |
| HSP | Heat shock protein |
| HSF1 | Heat shock factor 1 |
| IL | Interleukin |
| KynA | Kynurenic acid |
| LOX | Lysyl Oxidase |
| MMP | Matrix Metalloproteinase |
| PDGF | Platelet-derived growth factor |
| PGE2 | Prostaglandin E2 |
| TGFβ | Transforming growth factor beta |
| TME | Tumor microenvironment |
| TNF | Tumor necrosis factor |
| VEGF | Vascular endothelial growth factor |
References
- Nierengarten, M.B. Global cancer statistics 2022: The report offers a view on disparities in the incidence and mortality of cancer by sex and region worldwide and on the areas needing attention. Cancer 2024, 15, 2568. [Google Scholar] [CrossRef]
- Weth, F.R.; Hoggarth, G.B.; Weth, A.F.; Paterson, E.; White, M.P.; Tan, S.T.; Peng, L.; Gray, C. Unlocking hidden potential: Advancements, approaches, and obstacles in repurposing drugs for cancer therapy. Br. J. Cancer 2024, 130, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T cell therapy: A new era for cancer treatment. Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Chalbatani, G.M.; Jalali, S.A.; Mirzaei, H.R.; Grupp, S.A.; Suarez, E.R.; Rapôso, C.; Webster, T.J. CAR-T cells: Early successes in blood cancer and challenges in solid tumors. Acta Pharm. Sin. B 2021, 11, 1129–1147. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, B.; Pignatelli, C.; Cossutta, M.; Citro, A.; Courty, J.; Piemonti, L. The extracellular matrix in pancreatic cancer: Description of a complex network and promising therapeutic options. Cancers 2021, 13, 4442. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-cell therapy for solid tumors: Lessons learned from lymphoma treatment. Cancers 2020, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Leight, J.L.; Drain, A.P.; Weaver, V.M. Extracellular matrix remodeling and stiffening modulate tumor phenotype and treatment response. Annu. Rev. Cancer Biol. 2017, 1, 313–334. [Google Scholar] [CrossRef]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Ogando-Rivas, E.; Liu, R.; Wang, T.; Rubin, J.; Jin, L.; Tao, H.; Sawyer, W.W.; Mendez-Gomez, H.R.; Cascio, M.; et al. CAR T cell locomotion in solid tumor microenvironment. Cells 2022, 11, 1974. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Dandara, C. The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics 2023, 8, 146. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; del Río Hernández, A. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, E.; Bassi, D.E. Physico-mechanical aspects of extracellular matrix influences on tumorigenic behaviors. Semin. Cancer Biol. 2010, 20, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Mierke, C.T. The matrix environmental and cell mechanical properties regulate cell migration and contribute to the invasive phenotype of cancer cells. Rep. Prog. Phys. 2019, 82, 064602. [Google Scholar] [CrossRef] [PubMed]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.J. The role of the extracellular matrix in cancer stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Libby, J.R.; Royce, H.; Walker, S.R.; Li, L. The role of extracellular matrix in angiogenesis: Beyond adhesion and structure. Biomater. Biosyst. 2024, 15, 100097. [Google Scholar] [CrossRef]
- Kyriakou, G.; Melachrinou, M. Cancer stem cells, epigenetics, tumor microenvironment and future therapeutics in cutaneous malignant melanoma: A review. Future Oncology 2020, 16, 1549–1567. [Google Scholar] [CrossRef]
- Padhi, A.; Nain, A.S. ECM in differentiation: A review of matrix structure, composition and mechanical properties. Ann. Biomed. Eng. 2020, 48, 1071–1089. [Google Scholar] [CrossRef]
- Klabukov, I.; Smirnova, A.; Yakimova, A.; Kabakov, A.E.; Atiakshin, D.; Petrenko, D.; Shestakova, V.A.; Sulina, Y.; Yatsenko, E.; Stepanenko, V.N.; et al. Oncomatrix: Molecular Composition and Biomechanical Properties of the Extracellular Matrix in Human Tumors. J. Mol. Pathol. 2024, 5, 437–453. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.M.; He, Y. Exploring Extracellular Matrix Crosslinking as a Therapeutic Approach to Fibrosis. Cells 2024, 13, 438. [Google Scholar] [CrossRef] [PubMed]
- Denton, A.E.; Roberts, E.W.; Fearon, D.T. Stromal Cells in the Tumor Microenvironment. In Stromal Immunology. Advances in Experimental Medicine and Biology; Owens, B., Lakins, M., Eds.; Springer: Cham, Switzerland, 2018; pp. 99–114. [Google Scholar]
- Kabakov, A.E.; Yakimova, A.O. Hypoxia-induced cancer cell responses driving radioresistance of hypoxic tumors: Approaches to targeting and radiosensitizing. Cancers 2021, 13, 1102. [Google Scholar] [CrossRef]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Hapke, R.Y.; Haake, S.M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 2020, 487, 10–20. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, T.; Xia, R.; Wei, Y.; Wei, X. Targeting the tumor stroma for cancer therapy. Mol. Cancer 2022, 21, 208. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Smith, P.C. The dynamic interaction between extracellular matrix remodeling and breast tumor progression. Cells 2021, 10, 1046. [Google Scholar] [CrossRef] [PubMed]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-associated fibroblasts: Tumorigenicity and targeting for cancer therapy. Cancers 2022, 14, 3906. [Google Scholar] [CrossRef] [PubMed]
- Belhabib, I.; Zaghdoudi, S.; Lac, C.; Bousquet, C.; Jean, C. Extracellular matrices and cancer-associated fibroblasts: Targets for cancer diagnosis and therapy? Cancers 2021, 13, 3466. [Google Scholar] [CrossRef]
- Angioni, R.; Sánchez-Rodríguez, R.; Viola, A.; Molon, B. TGF-β in cancer: Metabolic driver of the tolerogenic crosstalk in the tumor microenvironment. Cancers 2021, 13, 401. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.F.; Jordan, P.; Matos, P. A signaling view into the inflammatory tumor microenvironment. Immuno 2021, 1, 91–118. [Google Scholar] [CrossRef]
- Pozzi, A.; Yurchenco, P.D.; Iozzo, R.V. The nature and biology of basement membranes. Matrix Biol. 2017, 57, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman Siveen, K.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.Q.; Uddin, S. Vascular endothelial growth factor (VEGF) signaling in tumour vascularization: Potential and challenges. Curr. Vasc. Pharmacol. 2017, 15, 339–351. [Google Scholar]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rey, D.; Carmona-Rodríguez, L.; Fernández-Aceñero, M.J.; Mira, E.; Mañes, S. Extracellular superoxide dismutase, the endothelial basement membrane, and the wnt pathway: New players in vascular normalization and tumor infiltration by T-cells. Front. Immunol. 2020, 11, 579552. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, J.; Li, L.; Liao, S.; He, J.; Zhou, S.; Zhou, Y. Pericytes in the tumor microenvironment. Cancer Lett. 2023, 556, 216074. [Google Scholar] [CrossRef] [PubMed]
- Boutilier, A.J.; Elsawa, S.F. Macrophage polarization states in the tumor microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Goswami, K.K.; Bose, A.; Baral, R. Macrophages in tumor: An inflammatory perspective. Clin. Immunol. 2021, 232, 108875. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.S.; Lund, A.W. Non-hematopoietic Control of Peripheral Tissue T Cell Responses: Implications for Solid Tumors. Front. Immunol. 2018, 9, 2662. [Google Scholar] [CrossRef] [PubMed]
- Gaimari, A.; De Lucia, A.; Nicolini, F.; Mazzotti, L.; Maltoni, R.; Rughi, G.; Zurlo, M.; Marchesini, M.; Juan, M.; Daniel Parras, D.; et al. Significant Advancements and Evolutions in Chimeric Antigen Receptor Design. Int. J. Mol. Sci. 2024, 25, 12201. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Lim, S.Y.; Kutikhin, A.G.; Gordon-Weeks, A.N. Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim. Biophys. Acta (BBA) Rev. Cancer 2018, 1870, 207–228. [Google Scholar] [CrossRef]
- Seoane, J.; Gomis, R.R. TGF-Β family signaling in tumor suppression and cancer progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef]
- Khan, E.S.; Däinghaus, T. HSP47 in human diseases: Navigating pathophysiology, diagnosis and therapy. Clin. Transl. Med. 2024, 14, e1755. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Yu, R.; Lian, F.; Zheng, Y.; Feng, S.; Li, C.; Zheng, X. Targeting HSP47 for cancer treatment. Anti-Cancer Drugs 2024, 35, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef]
- Singh, P.; Jay, D.G. The role of eHSP90 in extracellular matrix remodeling, tumor invasiveness, and metastasis. Cancers 2024, 16, 3873. [Google Scholar] [CrossRef]
- Singh, P.; Ramanathan, V.; Zhang, Y.; Georgakoudi, I.; Jay, D.G. Extracellular HSP90 binds to and aligns collagen-1 to enhance breast cancer cell invasiveness. Cancers 2023, 15, 5237. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Becker, J.C.; Andersen, M.H.; Schrama, D.; Straten, P.T. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol. Immunother. 2013, 62, 1137–1148. [Google Scholar] [CrossRef]
- Szöőr, A.; Tóth, G.; Zsebik, B.; Szabó, V.; Eshhar, Z.; Abken, H.; Vereb, G. Trastuzumab derived HER2-specific CARs for the treatment of trastuzumab-resistant breast cancer: CAR T cells penetrate and eradicate tumors that are not accessible to antibodies. Cancer Lett. 2020, 484, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. eBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef]
- van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef]
- Henze, J.; Tacke, F.; Hardt, O.; Alves, F.; Al Rawashdeh, W. Enhancing the Efficacy of CAR T Cells in the Tumor Microenvironment of Pancreatic Cancer. Cancers 2020, 12, 1389. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Y.; Chen, X. Collagen extracellular matrix promotes gastric cancer immune evasion by activating IL4I1-AHR signaling. Transl. Oncol. 2024, 49, 102113. [Google Scholar] [CrossRef] [PubMed]
- Niveria, K.; Yadav, M.; Dangi, K.; Verma, A.K. Overcoming challenges to enable targeting of metastatic breast cancer tumour microenvironment with nano-therapeutics: Current status and future perspectives. OpenNano 2022, 8, 100083. [Google Scholar] [CrossRef]
- Hong, M.; Talluri, S.; Chen, Y.Y. Advances in promoting chimeric antigen receptor T cell trafficking and infiltration of solid tumors. Curr. Opin Biotechnol. 2023, 84, 103020. [Google Scholar] [CrossRef]
- Zhu, C.; Wu, Q.; Sheng, T.; Shi, J.; Shen, X.; Yu, J.; Du, Y.; Sun, J.; Liang, T.; He, K.; et al. Rationally designed approaches to augment CAR-T therapy for solid tumor treatment. Bioact. Mater. 2023, 33, 377–395. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rodríguez, L.; Martínez-Rey, D.; Mira, E.; Mañes, S. SOD3 boosts T cell infiltration by normalizing the tumor endothelium and inducing laminin-α4. Oncoimmunology 2020, 9, 1794163. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-E.; Yun, S.; Doh, J. Effects of extracellular adhesion molecules on immune cell mediated solid tumor cell killing. Front Immunol. 2022, 13, 1004171. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.; Sebastian, A.; Hinckley, A.; Rios-Arce, N.D.; Hynes, W.F.; Edwards, S.A.; He, W.; Hum, N.R.; Wheeler, E.K.; Loots, G.G.; et al. Extracellular matrix modulates T cell clearance of malignant cells in vitro. Biomaterials 2022, 282, 121378. [Google Scholar] [CrossRef] [PubMed]
- Rømer, A.M.A.; Thorseth, M.-L.; Madsen, D.H. Immune Modulatory Properties of Collagen in Cancer. Front Immunol. 2021, 12, 791453. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Espinoza-Sánchez, N.A.; Götte, M. Role of cell surface proteoglycans in cancer immunotherapy. Semin. Cancer Biol. 2020, 62, 48–67. [Google Scholar] [CrossRef] [PubMed]
- Donelan, W.; Dominguez-Gutierrez, P.R.; Kusmartsev, S. Deregulated hyaluronan metabolism in the tumor microenvironment drives cancer inflammation and tumor-associated immune suppression. Front Immunol. 2022, 13, 971278. [Google Scholar] [CrossRef]
- Murdamoothoo, D.; Sun, Z.; Yilmaz, A.; Riegel, G.; Abou-Faycal, C.; Deligne, C.; Velazquez-Quesada, I.; Erne, W.; Nascimento, M.; Mörgelin, M.; et al. Tenascin-C immobilizes infiltrating T lymphocytes through CXCL12 promoting breast cancer progression. EMBO Mol. Med. 2021, 13, e13270. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Roberts, D.D. Emerging functions of thrombospondin-1 in immunity. Semin. Cell Dev. Biol. 2024, 155, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Prakash, J.; Shaked, Y. The Interplay between Extracellular Matrix Remodeling and Cancer Therapeutics. Cancer Discov. 2024, 14, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Tharp, K.M.; Kersten, K.; Maller, O.; Timblin, G.A.; Stashko, C.; Canale, F.P.; Menjivar, R.E.; Hayward, M.-K.; Berestjuk, I.; Hoeve, J.T.; et al. Tumor-associated macrophages restrict CD8+ T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat. Cancer 2024, 5, 1045–1062. [Google Scholar] [CrossRef]
- Park, J.A.; Cheung, N.-K.V. Promise and Challenges of T Cell Immunotherapy for Osteosarcoma. Int. J. Mol. Sci. 2023, 24, 12520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, H.; Li, F.; Huang, S.; Chen, F.; Li, Y. Bright future or blind alley? CAR-T cell therapy for solid tumors. Front Immunol. 2023, 14, 1045024. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Boluda, A.; Emmanuel Donnadieu, E. Obstacles to T cell migration in the tumor microenvironment. Comp. Immunol. Microbiol. Infect. Dis. 2019, 63, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Bruni, S.; Mercogliano, M.F.; Mauro, F.L.; Cordo Russo, R.I.; Schillaci, R. Cancer immune exclusion: Breaking the barricade for a successful immunotherapy. Front. Oncol. 2023, 13, 1135456. [Google Scholar] [CrossRef]
- Yin, X.; He, L.; Guo, Z. T-cell exhaustion in CAR-T-cell therapy and strategies to overcome it. Immunology 2023, 169, 400–411. [Google Scholar] [CrossRef]
- Zhang, J.; Li, J.; Hou, Y.; Lin, Y.; Zhao, H.; Shi, Y.; Chen, K.; Nian, C.; Tang, J.; Pan, L.; et al. Osr2 functions as a biomechanical checkpoint to aggravate CD8+ T cell exhaustion in tumor. Cell 2024, 187, 3409–3426.e24. [Google Scholar] [CrossRef]
- Martín-Otal, C.; Lasarte-Cía, A.; Serrano, D.; Casares, N.; Conde, E.; Navarro, F.; Sánchez-Moreno, I.; Gorraiz, M.; Sarrion, P.; Calvo, A.; et al. Targeting the extra domain A of fibronectin for cancer therapy with CAR-T cells. J. Immunother. Cancer 2022, 10, e004479. [Google Scholar] [CrossRef] [PubMed]
- Anil, J.; Alnemri, A.; Lytle, A.; Lockhart, B.; Anil, A.E.; Baumgartner, M.; Gebre, K.; McFerran, J.; Grupp, S.A.; Rheingold, S.R.; et al. Bone marrow fibrosis is associated with non-response to CD19 CAR T-cell therapy in B-acute lymphoblastic leukemia. Am. J. Hematol. 2023, 98, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Son, K.; Hwang, Y.; Ko, J.; Lee, Y.; Doh, J.; Jeon, N.L. High-Throughput Microfluidic 3D Cytotoxicity Assay for Cancer Immunotherapy (CACI-IMPACT Platform). Front Immunol. 2019, 10, 1133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Zhou, X.; Khan, E.S.; Alansary, D.; Friedmann, K.S.; Yang, W.; Schwarz, E.C.; Del Campo, A.; Hoth, M.; Qu, B. Targeting the Microtubule-Network Rescues CTL Killing Efficiency in Dense 3D Matrices. Front. Immunol. 2021, 12, 729820. [Google Scholar] [CrossRef] [PubMed]
- Joy, J.D.; Malacrida, B.; Laforêts, F.; Kotantaki, P.; Maniati, E.; Manchanda, R.; Annibaldi, A.; Hopkins, S.; Garrobo-Calleja, I.; Gautrot, J.; et al. Human 3D Ovarian Cancer Models Reveal Malignant Cell-Intrinsic and -Extrinsic Factors That Influence CAR T-cell Activity. Cancer Res. 2024, 84, 2432–2449. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, S.Y.; Killian, D.; Schulte, J.; Sticht, C.; Thiel, M.; Lindner, H.A. Short Term Hypoxia Synergizes with Interleukin 15 Priming in Driving Glycolytic Gene Transcription and Supports Human Natural Killer Cell Activities. J. Biol. Chem. 2016, 291, 12960–12977. [Google Scholar] [CrossRef] [PubMed]
- Hickey, J.W.; Dong, Y.; Chung, J.W.; Salathe, S.F.; Pruitt, H.C.; Li, X.; Chang, C.; Fraser, A.K.; Bessell, C.A.; Ewald, A.J.; et al. Engineering an Artificial T-Cell Stimulating Matrix for Immunotherapy. Adv. Mater. 2019, 31, e1807359. [Google Scholar] [CrossRef] [PubMed]
- Livingston, N.K.; Hickey, J.W.; Sim, H.; Salathe, S.F.; Choy, J.; Kong, J.; Silver, A.B.; Stelzel, J.L.; Omotoso, M.O.; Li, S.; et al. In Vivo Stimulation of Therapeutic Antigen-Specific T Cells in an Artificial Lymph Node Matrix. Adv. Mater. 2024, 23, e2310043. [Google Scholar] [CrossRef]
- Hu, Q.; Li, H.; Archibong, E.; Qian Chen, Q.; Ruan, H.; Ahn, S.; Dukhovlinova, E.; Kang, Y.; Wen, D.; Dotti, G.; et al. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat. Biomed. Eng. 2021, 5, 1038–1047. [Google Scholar] [CrossRef]
- Mohaghegh, N.; Ahari, A.; Zehtabi, F.; Buttles, C.; Davani, S.; Hoang, H.; Tseng, K.; Zamanian, B.; Khosravi, S.; Daniali, A.; et al. Injectable hydrogels for personalized cancer immunotherapies. Acta Biomater. 2023, 172, 67–91. [Google Scholar] [CrossRef]
- Liu, L.; Qu, Y.; Cheng, L.; Yoon, C.W.; He, P.; Monther, A.; Guo, T.; Chittle, S.; Wang, Y. Engineering chimeric antigen receptor T cells for solid tumour therapy. Clin. Transl. Med. 2022, 12, e1141. [Google Scholar] [CrossRef] [PubMed]
- Fucà, G.; Reppel, L.; Landoni, E.; Savoldo, B.; Dotti, G. Enhancing chimeric antigen receptor T-cell efficacy in solid tumors. Clin. Cancer Res. 2020, 26(11), 2444–2451. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Liu, B. Critical factors in chimeric antigen receptor-modified T-cell (CAR-T) therapy for solid tumors. Onco Targets Ther. 2018, 12, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Cui, Y.; Zheng, Y.; Li, S.; Lv, J.; Wu, Q.; Long, Y.; Wang, S.; Yao, Y.; Wei, W.; et al. Human Hyaluronidase PH20 Potentiates the Antitumor Activities of Mesothelin-Specific CAR-T Cells Against Gastric Cancer. Front. Immunol. 2021, 12, 660488. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Shen, K.; Liang, S.; Lyu, Y.; Zhang, S.; Dong, H.; Li, Y.; Han, Y.; Zhao, X.; Zhang, Y.; et al. Specific ECM degradation potentiates the antitumor activity of CAR-T cells in solid tumors. Cell Mol. Immunol. 2024, 21, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, S.; Li, S.; Qu, Y.; Wang, H.-Y.; Liu, J.; Dunn, Z.S.; Cinay, G.E.; MacMullan, M.A.; Hu, F.; et al. Secretion of bispecific protein of anti-PD-1 fused with TGF-β trap enhances antitumor efficacy of CAR-T cell therapy. Mol. Ther. Oncolytics 2021, 21, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Gu, A.; Bai, Y.; Zhang, C.; Xu, C.; An, Z.; Zhang, Y.; Zhong, S.H.; Hu, Y.; Zhong, X. IL13Rα2-targeted third-generation CAR-T cells with CD28 transmembrane domain mediate the best anti-glioblastoma efficacy. Cancer Immunol. Immunother. 2023, 72, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Wickman, E.; Lange, S.; Wagner, J.; Ibanez, J.; Tian, L.; Lu, M.; Sheppard, H.; Chiang, J.; Koo, S.C.; Vogel, P.; et al. IL-18R supported CAR T cells targeting oncofetal tenascin C for the immunotherapy of pediatric sarcoma and brain tumors. J. Immunother. Cancer 2024, 12, e009743. [Google Scholar] [CrossRef]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pei, P.; Zhou, H.; Xie, Y.; Yang, S.; Shen, W.; Hu, L.; Zhang, Y.; Liu, T.; Yang, K. Nattokinase-Mediated Regulation of Tumor Physical Microenvironment to Enhance Chemotherapy, Radiotherapy, and CAR-T Therapy of Solid Tumor. ACS Nano 2023, 17, 7475–7486. [Google Scholar] [CrossRef]
- Yang, X.; Li, C.; Yang, H.; Li, T.; Ling, S.; Zhang, Y.; Wu, F.; Liu, X.; Liu, S.; Fan, C.; et al. Programmed Remodeling of the Tumor Milieu to Enhance NK Cell Immunotherapy Combined with Chemotherapy for Pancreatic Cancer. Nano Lett. 2024, 24, 3421–3431. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kaur, A.; Pavlidaki, A.; Spenlé, C.; Rajnpreht, I.; Donnadieu, E.; Salomé, N.; Molitor, A.; Carapito, R.; Wack, F.; et al. Targeting the Matrix REgulating MOtif abolishes several hallmarks of cancer, triggering antitumor immunity. Proc. Natl. Acad. Sci. USA 2024, 121, e2404485121. [Google Scholar] [CrossRef] [PubMed]
- Suto, A.; Kudo, D.; Yoshida, E.; Nagase, H.; Suto, S.; Mimura, J.; Itoh, K.; Hakamada, K. Increase of Tumor Infiltrating γδ T-cells in Pancreatic Ductal Adenocarcinoma Through Remodeling of the Extracellular Matrix by a Hyaluronan Synthesis Suppressor, 4-Methylumbelliferone. Pancreas 2019, 48, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khalil, R.A. Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, L.; Su, H.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef]
- Hu, J.; Lazar, A.J.; Ingram, D.; Wang, W.-L.; Zhang, W.; Jia, Z.; Ragoonanan, D.; Wang, J.; Xia, X.; Mahadeo, K.; et al. Cell membrane-anchored and tumor-targeted IL-12 T-cell therapy destroys cancer-associated fibroblasts and disrupts extracellular matrix in heterogenous osteosarcoma xenograft models. J. Immunother. Cancer 2024, 12, e006991. [Google Scholar] [CrossRef]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fang, L.; Wang, X.; Yuan, S.; Li, W.; Tian, W.; Chen, J.; Zhang, Q.; Zhang, Y.; Zhang, Q.; et al. Oncolytic adenovirus-mediated expression of decorin facilitates CAIX-targeting CAR-T therapy against renal cell carcinoma. Mol. Ther. Oncolytics 2021, 24, 14–25. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Chung, C.-L.; Hu, T.-H.; Chen, J.-J.; Liu, P.-F.; Chen, C.-L. Recent progress in TGF-β inhibitors for cancer therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef] [PubMed]
- Chitty, J.L.; Setargew, Y.F.; Cox, T.R. Targeting the lysyl oxidases in tumour desmoplasia. Biochem. Soc. Trans. 2019, 47, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- Tempest, R.; Guarnerio, S.; Maani, R.; Cooper, J.; Peake, N. The biological and biomechanical role of transglutaminase-2 in the tumour microenvironment. Cancers 2021, 13, 2788. [Google Scholar] [CrossRef]
- Watanabe, N.; McKenna, M.K.; Rosewell Shaw, A.; Suzuki, M. Clinical CAR-T Cell and Oncolytic Virotherapy for Cancer Treatment. Mol. Ther. 2021, 29, 505–520. [Google Scholar] [CrossRef]
- Rezaei, R.; Esmaeili Gouvarchin Ghaleh, H.; Farzanehpour, M.; Dorostkar, R.; Ranjbar, R.; Bolandian, M.; Nodooshan, M.M.; Alvanegh, A.G. Combination therapy with CAR T cells and oncolytic viruses: A new era in cancer immunotherapy. Cancer Gene Ther. 2022, 29, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Uche, I.K.; Kousoulas, K.G.; Rider, P.J.F. The Effect of Herpes Simplex Virus-Type-1 (HSV-1) Oncolytic Immunotherapy on the Tumor Microenvironment. Viruses 2021, 13, 1200. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, M.; Qiu, G.; Rong, C.; Zhu, X.; Qin, G.; Kong, C.; Zhou, J.; Xiayi Liang, X.; Bu, Z.; et al. Extracellular Matrix Viscosity Reprogramming by In Situ Au Bioreactor-Boosted Microwavegenetics Disables Tumor Escape in CAR-T Immunotherapy. ACS Nano 2023, 17, 5503–5516. [Google Scholar] [CrossRef]
- Chen, Z.; Pan, H.; Luo, Y.; Yin, T.; Zhang, B.; Liao, J.; Wang, M.; Tang, X.; Huang, G.; Deng, G.; et al. Nanoengineered CAR-T Biohybrids for Solid Tumor Immunotherapy with Microenvironment Photothermal-Remodeling Strategy. Small 2021, 17, e2007494. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klabukov, I.; Kabakov, A.E.; Yakimova, A.; Baranovskii, D.; Sosin, D.; Atiakshin, D.; Ignatyuk, M.; Yatsenko, E.; Rybachuk, V.; Evstratova, E.; et al. Tumor-Associated Extracellular Matrix Obstacles for CAR-T Cell Therapy: Approaches to Overcoming. Curr. Oncol. 2025, 32, 79. https://doi.org/10.3390/curroncol32020079
Klabukov I, Kabakov AE, Yakimova A, Baranovskii D, Sosin D, Atiakshin D, Ignatyuk M, Yatsenko E, Rybachuk V, Evstratova E, et al. Tumor-Associated Extracellular Matrix Obstacles for CAR-T Cell Therapy: Approaches to Overcoming. Current Oncology. 2025; 32(2):79. https://doi.org/10.3390/curroncol32020079
Chicago/Turabian StyleKlabukov, Ilya, Alexander E. Kabakov, Anna Yakimova, Denis Baranovskii, Dmitry Sosin, Dmitry Atiakshin, Michael Ignatyuk, Elena Yatsenko, Victoria Rybachuk, Ekaterina Evstratova, and et al. 2025. "Tumor-Associated Extracellular Matrix Obstacles for CAR-T Cell Therapy: Approaches to Overcoming" Current Oncology 32, no. 2: 79. https://doi.org/10.3390/curroncol32020079
APA StyleKlabukov, I., Kabakov, A. E., Yakimova, A., Baranovskii, D., Sosin, D., Atiakshin, D., Ignatyuk, M., Yatsenko, E., Rybachuk, V., Evstratova, E., Eygel, D., Kudlay, D., Stepanenko, V., Shegay, P., & Kaprin, A. D. (2025). Tumor-Associated Extracellular Matrix Obstacles for CAR-T Cell Therapy: Approaches to Overcoming. Current Oncology, 32(2), 79. https://doi.org/10.3390/curroncol32020079